Congenital Adrenal Hyperplasia: Female Pseudohermaphroditism and Virilization
Authors
INTRODUCTION
Excessive androgen levels in the female may affect primary sexual characteristics by masculinizing the fetal external genitalia to produce female pseudohermaphroditism or secondary sexual characteristics by inducing changes similar to those characteristic of male puberty to produce virilization. Therefore, female pseudohermaphroditism is a condition in which the gonadal sex is female, but the external genitalia are masculinized. Ambiguous female genitalia occur only when excessive androgens are present during fetal life, and the degree of masculinization is dependent on the timing of exposure to these androgens. If the fetus is exposed after the 12th week, when the vagina and urethra have separated, only clitoral hypertrophy occurs. If androgen levels are high before then, labial fusion and a urogenital sinus result.
Virilization in the female is a condition characterized by any of the following: premature pubarche, hirsutism, acne, male-pattern baldness, or menstrual irregularities. It requires postnatal androgen exposure. Patients with masculinization frequently go on to virilize, but not all patients with virilizing syndromes have been masculinized, except perhaps for varying degrees of postnatal clitoral hypertrophy.
High androgen levels are most commonly caused by hyperplasia of the adrenal gland as a result of the genetic defects of steroidogenesis, called the virilizing congenital adrenal hyperplasias (CAHs).
ADRENAL FUNCTION
The adrenal cortex produces both corticosteroids (c21 steroids), which consist of the mineralocorticoids and glucocorticoids, and sex steroids, predominantly androgens (c19 steroids). The initial step in steroidogenesis(Fig. 1) is the conversion of cholesterol, a c27 sterol, by cholesterol desmolase (or cholesterol side-chain cleavage enzyme) to the c21 precursor pregnenolone. In the adrenal cortex, glucocorticoids and androgens are synthesized predominantly through the Δ5 pathway, in contrast to steroidogenesis in the gonad, which proceeds through progesterone along the Δ4 pathway. Thus, in the adrenal gland, pregnenolone is hydroxylated at the 17α position to 17α-hydroxypregnenolone, which undergoes conversion by the enzyme complex 3β-hydroxysteroid dehydrogenase (3β-HSD)-Δ5,Δ4-isomerase to the corresponding 3-oxo,Δ4-steroid, 17α-hydroxyprogesterone (17-OHP). Further hydroxylation of 17-OHP, first by the enzyme 21-hydroxylase, producing the intermediate 11-deoxycortisol, and then at the 11β position by 11β-hydroxylase, yields cortisol.
|
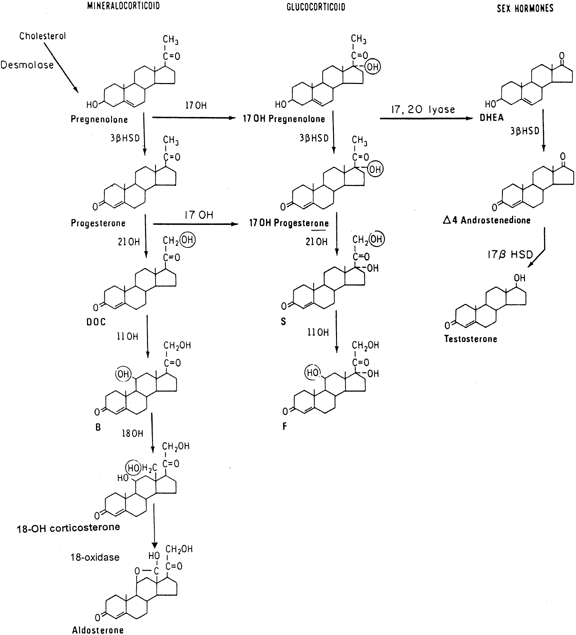
In the mineralocorticoid pathway, after cleavage of pregnenolone from cholesterol and direct oxidation by 3β-HSD-Δ5,Δ4-isomerase, progesterone is similarly hydroxylated at the 21 and 11β positions, yielding deoxycorticosterone and corticosterone. Two additional conversions of corticosterone, namely 18-hydroxylation and 18-dehydrogenation, both dependent on the same enzyme that catalyzes 11β-hydroxylation, are required for the completion of aldosterone synthesis. Thus, corticosterone is 18-hydroxylated to 18-hydroxycorticosterone, and further oxidation of the 18-hydroxyl group yields aldosterone.
The steroidogenic cells of the adult adrenal cortex may be divided into three histologically distinct zones: the zona glomerulosa, the zona fasciculata, and the zona reticularis. Biochemically, the 18-dehydrogenation step completing aldosterone synthesis occurs only in the outer zone, the zona glomerulosa. Conversely, the enzyme 17α-hydroxylase, which conducts both the 17α-hydroxylation necessary for glucocorticoid synthesis and the 17,20-lyase activity necessary for adrenal androgen biosynthesis, is active in the zona fasciculata and the zona reticularis.
The adrenal androgens dehydroepiandrosterone (DHEA) and Δ4-androstenedione (Δ4-A) are formed from the c21 precursor steroids 17α-hydroxypregnenolone and 17-OHP by removal of the remaining short side chain at position 17 by the 17,20-lyase activity. DHEA is readily converted to Δ4-A by the action of the enzyme 3β-HSD.
The main androgen secreted by the normal adrenal cortex is DHEA, which has little effect in itself, but may undergo peripheral conversion to steroids of greater androgenic potency. During adrenarche in late childhood, the first of a progression of hormonal changes culminating in puberty, sexual maturation, and the end of growth in stature, there commences a steady rise in the adrenal output of DHEA, which peaks in the early 20s.
In abnormal steroidogenesis from defects of the 21- and 11β-hydroxylase and 3β-HSD enzymes, accumulating cortisol precursors are channeled into unimpaired c19 pathways, increasing adrenal secretion of DHEA and Δ4-A. In clinically significant defects of these enzymes, the excess adrenal androgen secretion may alter external genital differentiation in gonadal females in fetal life, resulting in female pseudohermaphroditism;if untreated postnatally, this excess secretion leads to progressive virilization.
ADRENAL STEROIDS IN DEVELOPMENT
Fetal Genital Differentiation
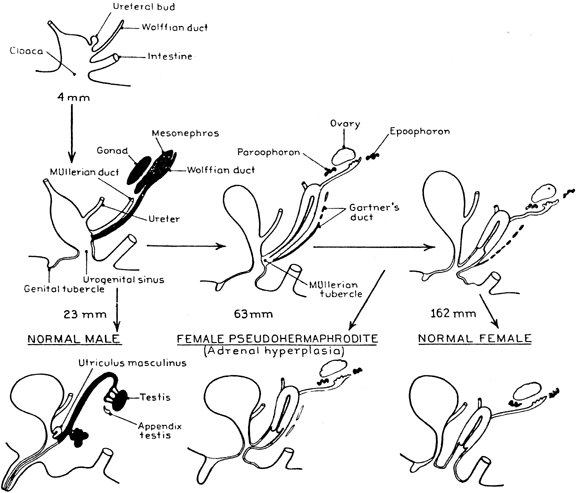
The fundamental development of the external genitalia in the fetus is female. Any masculinization, normal or pathologic, is the result of androgen action. Adrenocortical cell differentiation occurs early in embryogenesis, with the formation of a provisional fetal zone, active for the remainder of gestation that involutes after birth. Genital differentiation from the 8th to the 13th week in the fetus takes place in the presence of steroids synthesized by an already active adrenal cortex. Excessive androgen production during this critical period will cause masculinization of the external genitalia in a fetus of female gonadal sex. The extent of masculinization ranges from mild clitoral enlargement through varying degrees of fusion of the labioscrotal folds, to the profound morphologic anomaly of a penile urethra, which is very rare.
Male internal genital differentiation proceeds under the control of two hormonal factors produced in effective amounts only by the fetal testes: (1) testosterone, which directs formation of the male genital structures from the wolffian (mesonephric) ducts,1 and (2) the glycoprotein anti-müllerian müllerian hormone, which suppresses development of the müllerian ducts into the female internal structures (Fig. 2).2 Genetic sex and gonadal differentiation are normal in patients with CAH.3 Because there is no anomalous secretion of anti-müllerian hormone in females with CAH, the fallopian tubes, uterus, and upper vagina develop normally. Wolffian duct stabilization and differentiation require the high local androgen levels provided by the male gonads; androgen elevations of adrenal origin appear not to affect this process, and there is no observable wolffian duct development in females suffering even the most extreme virilization from androgen excess. Thus, internal genital morphogenesis is normal.
|
ADRENAL ENZYME DEFECTS ASSOCIATED WITH FEMALE PSEUDOHERMAPHRODITISM AND VIRILIZATION
The following enzymatic defects of steroidogenesis may cause female pseudohermaphroditism or virilization4,5,6:
- 3β-Hydroxysteroid dehydrogenase deficiency (classic and nonclassic CAH)
- 21-Hydroxylase deficiency (salt-wasting, simple virilizing, and nonclassic CAH)
- 11β-Hydroxylase (hypertensive classic and nonclassic CAH)
These syndromes are discussed in detail later in this chapter.
21-Hydroxylase Deficiency
CAH is the most common cause of female pseudohermaphroditism and virilization, and decreased cortisol synthesis owing to reduction or loss of 21-hydroxylase enzyme function is the most common biochemical cause of CAH.7,8,9 The decreased plasma cortisol elevates adrenocorticotropic hormone (ACTH) secretion,10,11,12 stimulating increased adrenal production of cortisol and of the androgen precursors and androgens, which do not require 21-hydroxylase for their biosynthesis.
Early clinical studies13 showed increased urinary levels of pregnanetriol, the principal metabolite of 17-OHP, and also of the 17-ketosteroids, metabolites of the androgens DHEA and Δ4-A, and testosterone, in patients with 21-hydroxylase deficiency. Determinations of serum levels of 17-OHP and Δ4 by radioimmunoassay allow more accurate diagnosis of CAH than could be provided formerly by the assessment of 24-hour urinary levels of hormonal metabolites.14,15
CLASSIC 21-HYDROXYLASE DEFICIENCY.
The prominent feature of 21-hydroxylase deficiency is progressive virilization with advanced somatic development. The classic disorder produces ambiguous or masculine external genital formation in female infants at birth. It occurs in two major forms, simple virilizing and salt-wasting. Because a salt-wasting crisis within the first few weeks of life can have profound effects on the infants, leading even to death, every infant born with ambiguous genitalia must have a prompt, thorough evaluation to exclude the salt-wasting forms of CAH.
Simple Virilizing Form
Developmental genital anomalies are manifest in females as varying degrees of genital ambiguity, which should alert the physician to the presence of the condition. CAH caused by 21-hydroxylase deficiency is the most common cause of ambiguous genitalia in the newborn female, and because affected females have the capacity for an entirely normal female sex role, including childbearing, it is very important to recognize this disorder in newborns with ambiguous genitalia. Without treatment, there is progressive virilization and early fusion of the epiphyses with resulting short stature.
Salt-Wasting Form
In 75% of patients with classic 21-hydroxylase deficiency, salt wasting occurs in infancy and is life-threatening. It is characterized by hyponatremia, hyperkalemia, inappropriate natriuresis, and low serum and urinary aldosterone with concomitantly high plasma renin activity (PRA). The increase in the proportion of salt-wasting cases in recent years may be attributed in part to advances in diagnostic techniques as well as to increased survival because of the availability of exogenous mineralocorticoid supplements.
Salt wasting is caused by an enzyme deficiency that significantly impairs aldosterone synthesis, and low levels of this essential mineralocorticoid result in inadequate sodium retention by the renal distal tubule. It may also result from the effect of certain hormonal precursors, thought to be mineralocorticoid antagonists, found in increased levels in 21-hydroxylase deficiency. Salt wasting may be particularly prominent in infancy, given the marginally competent sodium-conserving mechanism of the immature newborn renal tubule, and may lessen with age.16,17,18 Careful monitoring of PRA in patients will aid in determining their changing dietary sodium, glucocorticoid, and mineralocorticoid requirements.
The extent of virilization may be the same in simple virilizing and salt-wasting CAH. Thus, even mildly virilized infants with 21-hydroxylase deficiency should be observed carefully for signs of a potentially life-threatening crisis within the first few weeks after birth.
Glucocorticoids are essential for the normal development and functioning of the adrenal medulla. A recent study has shown that CAH compromises both the development and functioning of the adrenomedullary system.19 A 40%to 80% reduction of plasma epinephrine and metaepinephrine concentrations was found in patients with CAH. This reduction is probably caused by a combination of the lack of intraadrenal cortisol secretion and abnormal adrenomedullary formation.
Until recently, the literature has indicated that with few exceptions,20 presence or absence of salt wasting is consistent within a family, and it has been predicted that subsequent affected offspring will have the same form of the disease as the index case. However, several families have been reported in which concordance for salt wasting was lacking among human leukocyte antigen (HLA)-identical siblings16 (see later discussion on HLA and 21-hydroxylase). In some cases, differences in aldosterone-synthesizing capacity could also be demonstrated between affected siblings as well as in unrelated individuals carrying identical mutations.
NONCLASSIC 21-HYDROXYLASE DEFICIENCY.
An attenuated form of adrenal hyperplasia was first suspected in the early 1950s by gynecologists in clinical practice who used glucocorticoids to treat women with physical signs of hyperandrogenism, including infertility.21,22 The first documentation of suppression of 21-hydroxylase precursors in the urine of such women after glucocorticoid therapy was by Decourt and co-workers23 in 1957. During the next two decades, the empiric use of glucocorticoids for the treatment of virilized women became commonplace, as adrenal androgens were often assumed to be elevated in those patients. Development of a radioimmunoassay for 17-OHP in the 1970s made it possible to diagnose 21-hydroxylase defects by measuring serum elevations of this index compound (the principal substrate for 21-hydroxylase in the adrenal zona fasciculata).24 The autosomal-recessive mode of genetic transmission of the nonclassic form of 21-hydroxylase deficiency became apparent through family studies of classic 21-hydroxylase deficiency.25,26,27
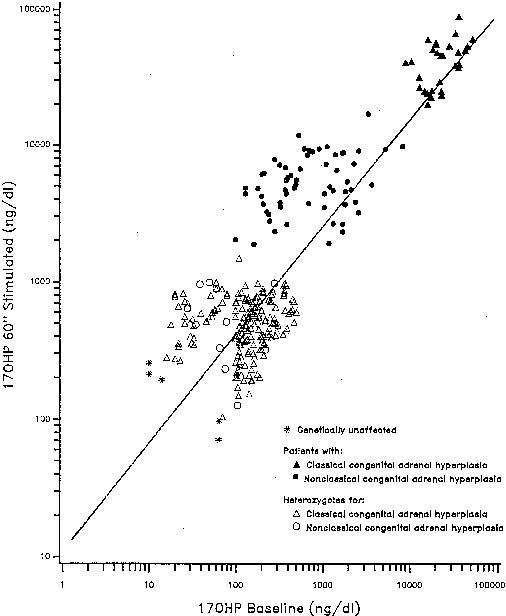
Individual 21-hydroxylase genotypes are revealed by the 17-OHP response to ACTH stimulation testing. Serum concentration samples are obtained at 0 (baseline) and 60 minutes after ACTH administration, with concentrations plotted on a reference nomogram (Fig. 3). The establishment of linkage to HLA28,29 confirmed the existence of this disorder as an allele of classic 21-hydroxylase deficiency.26,30 The HLA associations for nonclassic 21-hydroxylase deficiency28,29,31,32 are distinct from those found in classic 21-hydroxylase deficiency and differ according to ethnicity.29,33
|
Clinical Features
Clinical symptomatology of nonclassic 21-hydroxylase deficiency is variable;symptoms may appear at any age (Fig. 4), and longitudinal follow-up shows they may wax and wane with time. Certain patients hormonally identified as affected with nonclassic 21-hydroxylase deficiency are entirely asymptomatic up until the time of detection (usually as part of a family study). It is now thought, however, that emergence of overt hyperandrogenism is to be expected at some point in almost all cases.
|
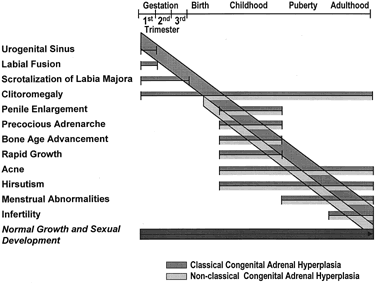
Nonclassic 21-hydroxylase deficiency can result in premature development of pubic hair in children; to our knowledge, the youngest such patient was noted to have pubic hair at 6 months of age.27 Elevated adrenal androgens promote the early fusion of epiphyseal growth plates, and it is commonly found that children with the disorder have advanced bone age and accelerated linear growth velocity and are ultimately shorter than the height that might be predicted on the basis of mid-parental height.34
Severe cystic acne refractory to oral antibiotics and retinoic acid has been attributed to nonclassic 21-hydroxylase deficiency. In addition, male-pattern baldness in young women with this disorder has been noted as the sole presenting symptom.
Menarche may be normal or delayed, and secondary amenorrhea is a frequent occurrence. Of patients with polycystic ovary syndrome, there is a subgroup of women with nonclassic 21-hydroxylase deficiency. The pathophysiology of this phenomenon probably relates to adrenal sex-steroid excess disrupting the usual cyclicity of gonadotropin release or the direct effects of adrenal androgens on the ovary, leading ultimately to the formation of ovarian cysts, which may then autonomously produce androgens.
Chronic hypersecretion of androgen precursors can induce a reduction in insulin sensitivity in female patients with nonclassic 21-hydroxylase deficiency.35
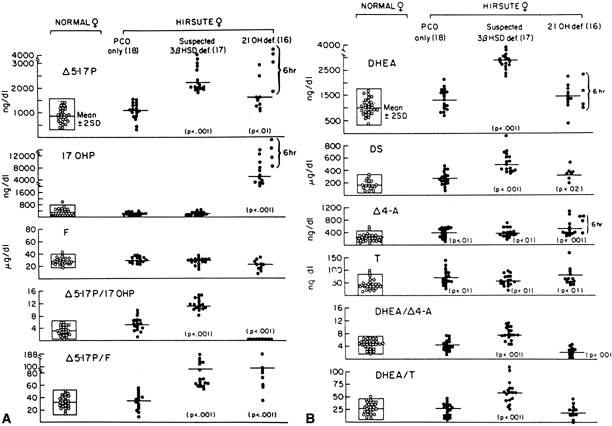
Although the androgen profile in serum and urine in both the basal and ACTH-stimulated states in this syndrome may not be markedly different from that demonstrated by women with polycystic ovary syndrome, the response of 17-OHP to ACTH clearly identifies those patients who have an adrenal 21-hydroxylase defect.36 Figure 5 presents nomograms for establishing the diagnosis of 21-hydroxylase deficiency (see later discussion on nonclassic 3β-HSD deficiency). Even ultrasonograms of the ovary do not distinguish between women with excess androgens from polycystic ovary syndrome and women with nonclassic 21-hydroxylase deficiency, and ACTH tests are required for the differential diagnosis. The response of the hypothalamic-pituitary-gonadal axis to luteinizing hormone-releasing hormone is variably abnormal in virilized women with nonclassic 21-hydroxylase deficiency.37
|
|
Treatment with glucocorticoids is effective in suppressing adrenal androgen production, and with time, clinical signs of androgen excess show improvement. Given the 9-month life expectancy of established hair follicles, remission of hirsutism generally takes at least 1 to 2 years. Because of the presumptive identification of the first nonclassic patients approximately 30 years ago, it has been recognized that infertility in women may be reversed during glucocorticoid therapy.21,22,23,38
Variability of Symptoms
The virilizing signs of nonclassic 21-hydroxylase deficiency are extremely variable among patients despite similar androgen levels. Thus, some patients with nonclassic 21-hydroxylase deficiency develop acne; others develop hirsutism, oligomenorrhea, or even reduced fertility; and some remain asymptomatic. The basis for the specific and idiosyncratic organ response to excess androgen remains unexplained. There may be interindividual variability in receptor sensitivity; alternatively, there may be both interindividual and intraindividual variation over time in the peripheral metabolism of skin or hair follicles. The disorder may be progressive, as demonstrated by the increasing prevalence of hirsutism with age that is observed among female patients with nonclassic CAH.39
Ultrasonography frequently demonstrates cystic ovaries, similar to those found in polycystic ovary syndrome, in patients with CAH caused by defects in 21-hydroxylase and other enzymes, the loss of which leads to a low level of cortisol. Will the suppression of adrenal androgens, readily accomplished in the treatment of CAH, cause reversal of the cystic changes in the ovary?Preliminary evidence indicates that this is likely, suggesting that cystic changes of the ovary in humans can result from excess androgens from adrenal or other extraovarian sources. Further, it may be valuable to test all women with cystic ovaries seen on ultrasonography for inborn errors of steroidogenesis, especially those women from ethnic groups at high risk for nonclassic 21-hydroxylase deficiency.
High Prevalence of 21-Hydroxylase Deficiency
The most common cause of female pseudohermaphrodi tism is CAH caused by 21-hydroxylase deficiency; both the classic and nonclassic forms of the disorder occur with high frequency in the populations that have been studied.
Neonatal screening tests suggest an incidence of 1 in 15,000 in most white populations for classic 21-hydroxylase deficiency.40 This disorder is thus relatively common for an inborn error of metabolism. Phenylketonuria, the most common of the inherited diseases for which neonatal screening is mandated, occurs in approximately 1 in 15,000 births in white populations.
The nonclassic form of 21-hydroxylase deficiency is even more common and is probably the most frequent autosomal-recessive genetic disorder in humans. Its incidence is especially high in Ashkenazi Jews (5%), Hispanics (2%), Yugoslavians (1.4%), and Italians (0.3%).33 In the diverse white population, the incidence is at least 0.1%.33,41,42
HUMAN LEUKOCYTE ANTIGENS AND 21HYDROXYLASE DEFICIENCY.
The genetic region called human leukocyte antigen (HLA), which is a part of the major histocompatibility complex (MHC), is a cluster of genes located on the short arm of chromosome 6. The class I antigens (HLA-A, -B, and -C), expressed on all nucleated cells, are the major barriers for allogenic transplantation. The class II antigens (principally HLA-D and -DR), expressed primarily on activated T-lymphocytes, participate in immune responses. The HLA complex also contains a linkage group of genes expressing products with immediate functions outside histocompatibility and immune response. The major components of class III antigens are the genes for C2 and C4 (C4a and C4b) of serum complement, factor B of the alternate complement pathway, and the 21-hydroxylase enzyme, cytochrome P450c21.
21-Hydroxylase deficiency is inherited as a monogenic autosomal-recessive trait closely linked to the HLA complex.43,44 Thus, with few exceptions,45 a sibling sharing both HLA haplotypes with the proband is predicted to be affected; one who shares a single haplotype is predicted to be heterozygote; and one who shares no HLA haplotype is predicted to be unaffected. De novo pathologic mutations are identified in those rare cases in which an HLA-identical sibling is not affected.46,47,48,49
HLA/21-Hydroxylase Linkage
The linkage between HLA and the 21-hydroxylase gene was first shown by Dupont and associates43 in a study on six families. A study of persons with intra-HLA recombinations suggested that the locus for 21-hydroxylase is situated between HLA-B and HLA-D, within the class III region.50,51
Linkage Disequilibrium
In addition to being linked to the adjacent HLA loci in general, 21-hydroxylase deficiency is found more frequently than expected with certain specific HLA antigens. Haplotypic associations (occurrence of genes linked on the same chromosome) in the HLA complex often include specific alleles of the other class III genes, complement components Bf, C2, and C4(C4A/B).52,53,54,55,56 Salt-wasting 21-hydroxylase deficiency is associated with HLA-Bw60 and with the extended haplotype HLA-A3;Bw47;DR7. This haplotype carries a null allele at locus C4B (thus expressing only one isotype of complement C4). Simple virilizing disease is associated with antigen HLA-Bw51 in selected ethnic groups, and the partial haplotype HLA-B14;DR1 is found to be associated with nonclassic disease in all ethnic groups examined except the Yugoslavian population. The B14;DR1 haplotype includes a duplication of one of the C4 loci.57 Another extended haplotype, HLA-A1,B8,DR3, is negatively associated with 21-hydroxylase deficiency; this hormonally normal haplotype, like the haplotype B47;DR7 (severe 21-hydroxylase deficiency), also expresses only one C4 isotype, in this case carrying a null allele at locus C4A.
Molecular Genetics
The structural gene encoding the adrenal cytochrome P450 specific for steroid 21-hydroxylation (P450c21) is named CYP21 or CYP21B and contains 10 exons.58,59,60 This gene and a 98% identical pseudogene (CYP21P or CYP21A) are located in close proximity (30 kb) in the HLA complex adjacent to and alternating with the C4B and C4A genes encoding the fourth component of the serum complement.61,62
The pseudogene CYP21P does not produce a detectable mRNA or a protein, owing to several deleterious mutations. Mutations in CYP21 appear to be generated by either of two types of recombination mechanisms. Misalignment of the tandem C4A-CYP21P-C4B-CYP21 arrangement during meiosis leads to unequal crossing over, resulting in a complete deletion of a DNA segment, including C4B and CYP21. Alternatively, small deleterious mutations appear to be transferred from CYP21P to CYP21 in gene conversion events.63,64 The frequency of gene deletions in different ethnic groups ranges from 11%to 35%, and many of these are found in association with the haplotype HLA-B47;DR7.65
CORRELATION OF GENOTYPE WITH PHENOTYPE.
In general, mutant P450c21 enzymes carrying specific amino acid substitutions identified in patients with 21-hydroxylase deficiency display activities that correspond roughly to the clinical severity of the disease and to the associated biochemical abnormalities (Table 1).
Table 1.
|
| Mutation |
|
|
|
Gene | Exon/Intron | Name/Type | AA | Comments | Reference |
CYP21 | E1 | Insertion (conversion) | +L9 | Normal polymorphism | 66 |
| E1 | Nonsense mutation | W22X |
| 67 |
| E1 | Frameshift | W22 +1nt | Insertion of 1 nucleotide | 68 |
| E1 | Missense mutation (conversion) | P30L | NC phenotype | 69 |
| E1 | Missense mutation | P30Q | SW allele | 70 |
| E1 | Frameshift mutation | Y47Δ1nt | Deletion of thymidine at nt 141 leads to L51X | 71 |
| I1 | Aberrant splicing of intron 1 | W23X nt 295 A |
| 67 |
| E2 | Missense mutation | G90V | Spanish patient | 72 |
| I2 | Aberrant splicing of intron 2 | nt 387 G | Intron 2 splice donor site Chinese patient | 73 |
| I2 | Aberrant splicing of intron 2 (conversion) | nt 656 A/C | Part of intron (end 19 bases) retained in mRNA processing. Most frequent nondeleted allele | 74 |
| E3 | Nonsense mutation | Y97X |
| 75 |
| E3 | Missense mutation | P106L | NC allele | 76 |
| E3 | Eight-base deletion (conversion) | G110Δ8nt | Frameshift: 20-AA + stop | 66 |
| E4 | One-base deletion | C169Δ1nt | Frameshift | 77 |
| E4 | Missense mutation (conversion) | I172N | Affects anchoring in membrane | 78 |
| E5 | Missense mutation | G178A | SW allele | 72 |
| E5 | Three-base deletion | ΔE196 | Deletion of nucleotides 1158–1160 | 79 |
| E6 | Cluster (conversion) | I236N V237E* M239K* | *2 more charges added in region with multiple charged residues | 74 |
| E7 | Missense mutation (conversion) | V281L | Major NC mutation HLA-B14;DR1 associated | 80 |
| E7 | Missense mutation | V281G |
| 81 |
| E7 | Missense mutation | G291S | AA substitution | 76 |
|
|
|
| C |
|
|
|
|
| position |
|
|
|
| b398 | At position +9 of |
|
|
|
|
| intron (secondary |
|
|
|
|
| effect?) |
|
| E7 | Missense mutation | G291C |
| 72 |
| E7 | Missense mutation | L300F |
| 81 |
| E7 | Nonsense mutation | W302X | Finnish patient | 82 |
| E7 | Single base insertion (conversion) | F306 +1nt | Frameshift: +T at codon 305–7 | 83 |
| I7 | Loss of splice donor site at Intron 7 | nt 1784 G | Aberrant splicing Found in one SW patient | 84 |
| I7 | Loss of splice donor site at Intron 7 | nt 1785 T | Aberrant splicing Found in one NC patient | 85 |
| E8 | Nonsense mutation | R316X | Chinese patient | 73 |
| E8 | Nonsense mutation (conversion) | Q318X |
| 86 |
| E8 | Frameshift | S330 Δ10 nt | Chinese patient | 73 |
| E8 | Missense mutation | R339H | NC allele | 87 |
| E8 | Missense mutation | R354H | 0% activity in transfected cells | 72 |
| E8 | Missense mutation | R354C |
| 81 |
| E8 | Missense mutation | R356W | Radical AA | 88 |
|
| (conversion) |
| substitution |
|
|
|
|
| May impair redox interactions |
|
| E8 | Missense mutation | R356P | May impair redox | 89 |
|
|
|
| interactions |
|
| E8 | Missense mutation | R356Q | May impair redox | 89 |
|
|
|
| interactions |
|
| E9 | Missense mutation | E380D |
| 90 |
| E9 | Duplication | V397 +16nt | Frameshift Chinese patient | 73 |
| E9 | Nonsense mutation | W405X |
| 76 |
| E10 | Missense mutation | G424S | Brazilian patient | 91 |
| E10 | Missense mutation | P453S+ plus P105L | NC allele AA substitution of conserved (P453) and nonconserved (P105) residue | 76, 92 |
| E10 | Frameshift mutation | P475 Δ1nt |
| 85 |
| E10 | Missense mutation | R483P | Possible first step of 2-step mechanism generating no. 39 | 93 |
| E10 | Compound frameshift mutation | R483 Δ1nt | Replaces last 11 AA and extends protein by a further 45 AA | 76 |
 G
G
NC, Nonclassical.
*Nucleotide number.
Like a homozygous deletion that precludes the expression of any enzyme, deletion in conjunction with a stop mutation or with a cluster of mutations at exon 6, which confers zero enzyme activity in vitro, would be predicted to result in 0% overall 21-hydroxylase activity in vivo and the severe salt-wasting phenotype. Homozygosity for the mutation Ile-172-Asn, which confers approximately 2% of normal activity on the gene product, usually results in the simple virilizing phenotype. However, the distinction between the two forms of classic 21-hydroxylase deficiency is not absolute.
Speiser and associates94 classified 90 patients into three mutation groups based on the degree of predicted enzymatic compromise. Mutation group A (no enzymatic activity) consisted primarily of salt-wasting patients, group B (2% activity) of simple virilizing patients, and group C (10% to 20% activity) of nonclassic patients. Mutation groups were correlated with clinical diagnosis, but each group contained patients with phenotypes either more or less severe than predicted. The phenotype was accurately predicted in 87% (54/62) of group A, in 72% (16/22) of group B, and in 62.5% of group C. A recent study by Wilson and colleagues found that the 10 most common mutations observed in the 21-hydroxylase gene result in phenotypes that are not always concordant with genotype.95 Genetic heterogeneity has been found in all populations studied thus far.81,96,97,98,99,100,101,102,103,104,105
Thus, the phenotype of a patient cannot be predicted from the genotype with complete certainty. Patients with phenotypes that are more severe than predicted from the genotype and who are discordant with siblings may have additional, as yet unidentified, mutations within the CYP21 gene. It is also plausible that at least some differences in clinical disease expression are governed by factors remote from the CYP21 locus. One could postulate that phenotypic severity is influenced by parental imprinting or by negative allelic complementation (i.e., exaggerated gene dosage effect).106 Activity of other gene-encoding proteins other than P450c21 that have steroid 21-hydroxylase activity is another possibility to explain phenotypic heterogeneity.102 Finally, patients with CAH lacking mutations in the entire 21-hydroxylase gene have also been described.107
3 β -Hydroxysteroid Dehydrogenase Deficiency
In 3β-HSD deficiency, as in the other two common forms of CAH known to produce ambiguous genitalia in the newborn, there is a spectrum of clinical phenotypes, including both salt-wasting and non-salt-wasting forms.108 The degree of severity of the enzyme defect cannot be determined from the appearance of the external genitalia at birth.
A defect in 3β-HSD was first described by Bongiovanni109 in 1962. On the basis of pedigree analysis, a monogenic autosomal-recessive mode of inheritance seemed most likely.108,109,110 This disorder affects the synthesis of all classes of adrenocortical steroids. Deficiency of the 3β-HSD enzyme may be diagnosed by measuring elevated levels of the Δ5-steroids: pregnenolone, 17α-hydroxypregnenolone, and DHEA in serum, and pregnanetriol and 16-pregnanetriol in urine. An elevated ratio of Δ5 to Δ4 steroids characterizes the biochemical findings in patients with 3β-HSD deficiency.
Unlike 21-hydroxylase, this enzyme is active in the gonads as well as in the adrenal glands, and a deficiency of 3β-HSD may cause both male and female pseudohermaphroditism. The resulting androgen deficiency in affected males will usually cause some degree of hypospadias (often the severe perineal-scrotal form) and palpable testes. Affected females, although frequently normal, may have clitoromegaly from very high levels of the weak androgen DHEA, which may undergo peripheral conversion to more potent androgens. The deficiency of aldosterone in classic cases of 3β-HSD deficiency results in salt wasting.109,111,112,113,114 Many cases, however, have been described in which the ability to conserve sodium was intact.108,110,111,113,115,116,117,118,119,120,121,122,123
It is claimed that 3β-HSD deficiency is the second most common steroidogenic defect,124 but as yet there have been no epidemiologic studies. There have been no reports of geographic or ethnic predominance of the disorder. As is the case for 21-hydroxylase deficiency, an attenuated or nonclassic form of the disease appears more common than the severe deficiency.
Nonclassic 3 β -HSD Deficiency
Nonclassic 3β-HSD deficiency is usually identified in girls with premature adrenarche or in adolescent and young adult women with hirsutism, acne, and oligomenorrhea.36,125 Until recently, these young women were usually diagnosed as having polycystic ovary syndrome, but careful scrutiny of ACTH-stimulated adrenal hormone responses has allowed differentiation of this subgroup.36 Little is known about symptomatic effects in males; cases presenting clinically at a later age have all had some degree of hypospadias, indicating presence of an enzyme defect already in prenatal life.
Mild 3β-HSD deficiency appears to be a relatively common cause of hirsutism in women. Fifteen percent of women with hirsutism in one study had the disorder, compared with 14% with late-onset 21-hydroxylase deficiency;the rest had no diagnosable adrenal defect.36 The mean age at diagnosis was 20 years, and although the patients tended to be of normal stature, there was some indication that the more severely affected women experienced an early growth acceleration in childhood, but as adults were short for mid-parental height. They were not obese. They tended to have a somewhat early pubarche and thelarche; 13% of patients with premature pubarche in one study had mild 3β-HSD deficiency,126 but the age at menarche was normal (11.8 years). Hirsutism or acne had its onset at the time of menarche or shortly thereafter, and about half of the women had onset of oligomenorrhea at the time of, or within the next 2 years after, onset of hirsutism or acne. A recent study has excluded partial 3β-HSD deficiency among 78 hirsute women.127
The diagnosis of mild 3β-HSD deficiency requires the demonstration of abnormally elevated Δ5-17α-hydroxypregnenolone (Δ5-17P) and DHEA levels in response to a standard ACTH stimulation test.36 Especially helpful in the diagnosis are the ratios of Δ5-17P:17-OHP and Δ5-17P:cortisol, which are characteristically elevated in 3β-HSD deficiency, thus differentiating it from 21-hydroxylase deficiency (see Fig. 5A). Baseline gonadotropin levels tend to be in the normal early- to late-follicular range, regardless of menstrual disturbance or presence of ovarian cysts, although quite a few patients may manifest an elevated base line luteinizing hormone. Adrenal computed tomography (CT) and abdominal ultrasound, when adequately performed, unequivocally show mild to marked bilateral adrenal hyperplasia.
Molecular Genetics
The enzyme 3β-HSD has the compound function of 3β-hydroxysteroid dehydrogenation and 3-oxosteroid isomerization. Two genes encoding 3β-HSD mapped to the 1p13 chromosome have been cloned to date: a skin-placental form (type I) and an adrenal-gonadal form (type II). Studies on the type II 3&gr;-HSD gene from index cases have revealed a number of mutations associated with different phenotypic forms of 3β-HSD deficiency.128,129,130,131,132,133 Molecular analysis to date has not shown conclusively that DNA abnormality is present in patients with the nonclassic form of the disease,134 although a heterozygous mutation of the type II gene in one case135 demonstrated that it is likely.
11 β -HYDROXYLASE DEFICIENCY.
Eberlein and Bongiovanni136,137 were the first to describe a congenital disorder of steroid biosynthesis causing virilization and hypertension with a pattern of steroid secretion indicating defective 11β-hydroxylation. This form accounts for approximately 5% of all cases of CAH.7,8,9,138
Classic 11 β -Hydroxylase Deficiency
As in 21-hydroxylase deficiency, excess fetal androgen production causes prenatal virilization of females, resulting in ambiguous external genitalia with normal female internal reproductive organs. In newborn males with 11β-hydroxylase deficiency, the external genitalia may be normal, but in either sex, virilization ensues postnatally if the disorder is untreated.
Deficiency of 11β-hydroxylase results in rising levels of the 11-deoxysteroids, 11-deoxycortisol (compound S), and 11-deoxycorticosterone (DOC), a moderately potent salt-retaining steroid. The elevated DOC causes sodium retention, plasma volume expansion, and suppression of PRA. Indeed, suppressed PRA is considered a hallmark of this defect, but exceptions have been noted.139 If present, hypertension is the single clinical feature of this disorder that distinguishes it from 21-hydroxylase deficiency. Hypokalemia and alkalosis resulting from mineralocorticoid hormone excess are inconstant features of this form of CAH. It is not entirely clear whether DOC is the agent causing elevation of blood pressure.89 The presence of mineralocorticoid excess and hypertension is not necessarily proportional to the degree of hypokalemia, nor is there a direct correlation between the degree of virilization and hypertension.140 Deficiency of 11β-hydroxylase accounts for approximately 5% of the cases of CAH worldwide.
Molecular Genetics
Deficiency of 11β-hydroxylase activity is inherited in an autosomal recessive manner, but unlike 21-hydroxylase deficiency, it is not HLA linked.141,142,143 Distinct isoenzymes of the mitochondrial P450 enzyme P450c11 participate in cortisol and aldosterone synthesis in humans. These isoenzymes, encoded by two genes CYP11B1 and CYP11B2, are 93%identical and located on the long arm of chromosome 8.144,145
In the zona fasciculata, 11β-hydroxylation of 11-deoxycortisol yields cortisol in a single step. In the zona glomerulosa, P450c11 hydroxylates DOC to corticosterone and has 18-hydroxylase and 18-oxidase activities, converting corticosterone to 18-hydroxycorticosterone and then aldosterone.146 Errors in these steps can lead to aldosterone synthase deficiency, types I and II. In the zona fasciculata, there is some 18-hydroxylation of DOC and corticosterone, but no aldosterone synthesis. Studies of patients with defective cortisol or aldosterone synthesis caused by respective deficiencies in 11β-hydroxylase and aldosterone synthase type II activities have confirmed the hypothesis that the isoenzyme encoded by CYP11B1 synthesizes cortisol in the zona fasciculata, whereas the isoenzyme encoded by CYP11B2 synthesizes aldosterone in the zona glomerulosa.147,148 Mutations in the zona fasciculata gene result in defective cortisol synthesis and hypertension caused by elevated DOC levels, whereas mutations in the zona glomerulosa gene result in defective aldosterone synthesis and salt wasting. Many patients in the Moroccan-Israeli population share the same mutation, Arg-448-His, in CYP11B1, which encodes a defective enzyme.149 Among the number of known mutations,150,151,152,153,154,155,156 most are clustered in exons 6 to 8. This clustering may reflect the location of functionally important amino acid residues within the enzyme or an increased tendency to develop mutations within this region of the gene.
Nonclassic 11 β -Hydroxylase Deficiency
Mild, nonclassic, and even asymptomatic forms of 11β-hydroxylase deficiency have been reported157,158,159,160,161,162 and may represent allelic variants. Elevated serum 17-OHP:cortisol or 11-deoxycortisol:cortisol ratios are seen in some heterozygotes, but in one study of obligate heterozygote parents, no consistent biochemical defect could be established in the base line state or after ACTH stimulation.163 It appears that hormonal values for carriers of (presumed) mild-deficiency alleles overlap with the normal range.
If this disorder follows the model of 21-hydroxylase deficiency in which structural gene mutations have been correlated with disease, then it is anticipated that mutations in the 11β-hydroxylase structural gene can produce the spectrum of clinical symptoms seen for 11β-hydroxylase deficiency (hypertension and virilization) and for 18-hydroxylase deficiency and 18-dehydrogenase deficiency (salt wasting without virilization). Further work is required, however, to determine whether the clinical polymorphism results from genetic allelism.
OTHER SOURCES OF ANDROGENIC HORMONES
Neoplasms
Maternal adrenal neoplasms may be the source of masculinizing androgens during the critical period of sexual differentiation in a fetus and may result in female pseudohermaphroditism.164 Although ovarian tumors are more commonly responsible for this type of presentation,165 very few cases have been reported in the world literature, all implicating adrenocortical adenomas.166,167,168,169,170 A paradoxical feature of these cases is the relatively mild virilization manifest in the mothers, even during pregnancies with marked masculinization of the fetus. In one sense, it is not surprising that maternal virilization was found to be mild, because the adenoma would have had to escape detection before pregnancy and cause no interference with fertility. Moreover, in two of three cases, androgen secretion by the adenoma was shown to respond to human chorionic gonadotropin (hCG) stimulation, suggesting that placental hCG exacerbated the maternal hyperandrogenemic state.166,168,170 The spectrum of masculinization of the fetus is broad, from mild clitoromegaly to a complete penile urethra. Testosterone is not the principal androgen produced by these adrenal tumors; DHEA and DHEA sulfate (DHEA-S) predominate. Because these are weak androgens, it is possible that excessive maternal levels of DHEA and DHEA-S are activated to more potent androgens by the fetoplacental unit.
A fascinating, but even rarer, cause of female pseudohermaphroditism is fetal adrenal adenoma. Such a tumor was discovered in a female pseudohermaphrodite.171 At age 25 years, she had the onset of Cushing's syndrome and virilization, and an adrenal tumor was surgically removed. Because the evaluation for a cause of pseudohermaphroditism was unrevealing during childhood, it is tempting to assume that the tumor secreted significant amounts of androgens in utero and, thereafter, became dormant or secreted small amounts of androgens without clinical significance. At age 25 years, the biological behavior of the tumor changed again, resulting in Cushing's syndrome and virilization. That the two conditions are unrelated is also possible, but less likely. Only one other case has been reported in more than 30 years, a girl with clitoromegaly at birth who was found to have a congenital adrenal adenoma.172
Administered Steroids
Androgens, progestins (natural and synthetic), and estrogens administered to the mother during gestation all have been reported to masculinize the female fetus, presumably after transplacental passage, by the same mechanism assumed for endogenous androgenic steroids.173 Although a large number of compounds have been implicated, case reports of these occurrences have disappeared from the literature. This situation probably reflects the general recognition that all medications may have as yet unrecognized teratogenic potential when used during pregnancy.
Androgens implicated include testosterone, methyltestosterone, methandriol, and normethandrone. Because their masculinizing potential is now so widely appreciated, they are seldom or never used during pregnancy. Progestogens implicated include ethisterone, norethindrone (these two appear to be the most potent), norethynodrel, progesterone, 17-hydroxyprogesterone, and medroxyprogesterone. The incidence of female pseudohermaphroditism after administration of progestogens during gestation is actually quite low. Because the dose of progestogens in contraceptive pills is small, there is little risk of masculinization from that medication's being administered early in pregnancy. Masculinization depends both on the dose and potency of the agent and on the duration and timing of administration; the earlier an agent is used during the critical period of sexual differentiation, the more severe the degree of masculinization.
The only estrogen clearly implicated in female pseudohermaphroditism is diethylstilbestrol,174 which is now never administered during pregnancy because of its association with vaginal clear-cell adenocarcinoma of the vagina and cervix in female offspring.175 Considering the frequency with which estrogens have been administered to maintain pregnancy, the incidence of masculinization from estrogen therapy must be extremely low. The mechanism of masculinization is not clear.
The present scarcity of iatrogenic female pseudohermaphroditism should not make the practitioner any less vigilant. Administration of sex steroids to the mother during pregnancy is a well-established cause of pseudohermaphroditism in the female offspring.
TREATMENT
Hormonal Replacement Therapy
The fundamental aim of endocrine therapy for CAH is to provide replacement of the deficient hormones. Since 1950, when Wilkins and colleagues176 and Bartter177 discovered the efficacy of cortisone therapy for CAH from 21-hydroxylase deficiency, glucocorticoid therapy has been the keystone of treatment for this disorder.
Glucocorticoid administration both replaces the deficient cortisol and suppresses ACTH overproduction. With cessation of overactivity of the adrenal cortex, there is reduced production of other adrenal steroids and amelioration of their noxious effects. Adrenal suppression in 21- and 11β-hydroxylase deficiency and in 3β-HSD deficiency reduces production of androgens, averting further virilization, slowing accelerated growth and bone age advancement to a more normal rate, and allowing a normal onset of puberty. Suppression of adrenal activity in 11β-hydroxylase deficiency diminishes DOC secretion and often results in remission of hypertension. For patients with a 21-hydroxylase or 3β-HSD deficiency and impaired mineralocorticoid synthesis, the administration of a salt-retaining steroid is required to maintain adequate sodium balance. Excessive glucocorticoid administration should be avoided, as this produces cushingoid facies, growth retardation, and inhibition of epiphyseal maturation.
Hydrocortisone (cortisol) is most often used. It is a physiologic hormone and does not introduce the complication of adjustment for potency, biological half-life, or altered profile of steroid action. Oral administration is the preferred and usual mode of treatment. The dosage is conventionally given daily in divided doses: 10 to 20 mg/m2 per day hydrocortisone, usually divided equally in two daily doses by tablet form, is adequate for the otherwise healthy child. In non-life-threatening illness or stress, a dose increase of two to three times the maintenance regimen is indicated for a few days. Each family must be given injection kits of hydrocortisone (50 mg for young children, 100 mg for older patients) for emergency use. In the event of a surgical procedure, a total of 5 to 10 times the daily maintenance dose (depending on the nature of the operative procedure) may be required during the first 24 hours; the dosage can then be rapidly tapered.
If there is a poor response to hydrocortisone at the standard dose, the dosage may be increased to 20 to 30 mg/m2/day in divided doses, or the regimen may be changed to either one of the hormonal analogs prednisone or dexamethasone. These agents are more potent and are longer acting, although their relative glucocorticoid and mineralocorticoid effects differ and the smaller amounts used make dosage adjustment more critical.
Because of individual variations in hepatic capability for metabolizing 11-oxosteroids, and thus in plasma clearance and half-life, prednisolone (the 11β-hydroxy analog of prednisone) is found in some patients to be more effective than prednisone for the replacement of glucocorticoid. Patients with classic 21-hydroxylase deficiency and salt wasting require mineralocorticoid replacement. The cortisol analog 9α-fluorohydrocortisone (Florinef) is used for its potent mineralocorticoid activity. In an adrenal crisis, if the patient is unable to ingest medications, liberal infusions of isotonic saline as well as parenteral hydrocortisone, at a dosage of 100 mg/m2/day for its mineralocorticoid activity, should be used. It is unfortunate that because of its potency, parenteral mineralocorticoid deoxycorticosterone acetate is no longer available in the United States.
Although aldosterone levels are not deficient in simple virilizing 21-hydroxylase deficiency, it has long been recognized that PRA may be elevated in the absence of salt wasting.178,179,180,181,182,183,184 Rösler and associates140 found that PRA in 21-hydroxylase deficiency is closely correlated with the ACTH level. They demonstrated that the addition of a mineralocorticoid in the hormonal therapy for patients with simple virilizing CAH and elevated PRA normalizes PRA and improves hormonal control, often with a reduced glucocorticoid requirement. Normalization of PRA also resulted in improved statural growth in these patients, a finding that has been corroborated in subsequent reports.183,184
Serum 17-OHP and Δ4-A concentrations determined by radioimmunoassay provide a sensitive index of biochemical control in patients with 21-hydroxylase deficiency.185,186,187 In females and prepubertal males, but not in newborn and pubertal males, the serum testosterone level is also a useful index.186 The combined determinations of PRA, 17-OHP, and serum androgens, as well as the clinical assessment of growth and pubertal status, must all be considered in adjusting the dose of glucocorticoid and salt-retaining steroid for optimal therapeutic control. Both in our clinic and in others, combinations of hydrocortisone and 9α-fluorohydrocortisone have proved highly effective treatment modalities.185
Measurement of PRA can be used to monitor efficacy of treatment not only in 21-hydroxylase deficiency, but also in other salt-losing forms of CAH, (e.g., 3β-HSD deficiency). It is also useful as a therapeutic index in those forms of CAH with mineralocorticoid excess and suppressed PRA (e.g., 11β-hydroxylase deficiency). In cases of poor hormonal control, PRA is elevated in the salt-losing forms and suppressed in the mineralocorticoid excess forms. Although glucocorticoid treatment has been available since 1950, there is no agreement as to which regimen gives the best outcome in terms of height. Recent studies suggest that even the most compliant patient may not achieve a final height compatible with parental stature.34,188 Early diagnosis and good compliance may confer some advantage for adult height outcome. Is this poor prognosis for height caused by overtreatment or the failure of oral glucocorticoid therapy given twice or even three times daily to keep excess androgen production suppressed? A simple system is necessary for home monitoring the hormonal status of patients at sufficiently frequent intervals to ensure hormonal control. Using salivary 17-OHP concentrations may provide an easy means to monitor hormone levels on a daily basis.189 Similar considerations may apply to the preservation of fertility. In a recent study, the use of growth hormone alone and in combination with gonadotropin-releasing hormone analog in children with poor predicted final heights was found to decrease height deficit after 1 and 2 years of treatment.190 The authors believe a possible explanation for the excellent response to GH treatment in CAH patients may be that the excess adrenal androgen secretion, which is not completely suppressed by glucocorticoid treatment, acts synergistically with GH to cause growth acceleration.
Intersex Management
Sexual ambiguity at birth, a characteristic of female pseudohermaphroditism, in most cases requires a rapid, rational, and judicious choice of sex assignment. This is a critical aspect of treatment, because the decision of sex assignment has obvious lifelong implications. Determination of genetic sex by karyotype or buccal smear and the accurate diagnosis of the specific underlying enzymatic defect are essential in assessing a patient's potential for future sex role and fertility.
In cases of female pseudohermaphroditism resulting from 21- or 11β-hydroxylase deficiency or from 3β-HSD deficiency, a female sex assignment is appropriate. When medical treatment is begun early in life, the initially large and prominent clitoris shrinks slightly, and as the surrounding structures grow normally, it becomes much less prominent and surgical revision may not be required. When the clitoris is conspicuously enlarged or when the abnormal genitalia interfere with parent-child bonding, plastic surgery to correct the appearance of the clitoris should be carried out in infancy. Definitive vaginoplasty should, in general, be performed in early to mid-adolescence by an experienced gynecologic surgeon.191 Because of the normal internal genitalia and gonads in these patients, normal puberty, fertility, and childbearing are possible when there is early therapeutic intervention.192 In view of this potential for normal female sexual development, it is unfortunate when, as a result of a hasty delivery room examination of the virilized external genitalia, 21-OH deficient females are improperly assigned and reared as males.
In assigning the sex of rearing to a female pseudohermaphrodite, the genetic sex may be of less consideration than the physiologic and anatomic character of the genitalia, their potential for development and function, and the psychosocial milieu of the infant. Because of the wide individual variability in the presentation of ambiguous genitalia in these patients, there can be no all-inclusive rules for sex assignment based solely on genetic sex or type of enzyme deficiency. Nonetheless, it is quite unusual currently for a female pseudohermaphrodite with one of the CAH syndromes to be raised as a male.
Psychologists and psychiatrists well acquainted with these endocrine disorders provide a vital component of the treatment regimen, because one of the major goals of therapy is to ensure that gender role, gender behavior, and gender identity are isosexual with the sex of assignment.193
That steroids influence aspects of central nervous system development and function is well recognized,194,195 but there are controversial data with regard to specific androgen effects resulting from CAH. It has been proposed that a masculinized gender role in girls, with behavioral manifestations such as tomboyishness, is a result of prenatal androgen excess in CAH. Similarly considered, behavioral changes resulting from other alterations in the androgen milieu are seen in studies on male pseudohermaphrodites with 5α-reductase or 17-ketosteroid reductase deficiency, who often elect a male gender identity at puberty.196,197 Yet, there is a great body of careful psychological studies indicating that in humans, unlike other mammals, the sex of rearing overrides prenatal hormonal effects.198
Prenatal Diagnosis and Treatment
Prenatal diagnosis and treatment are possible in pregnancies at risk for CAH. Prenatal diagnosis of 21-hydroxylase deficiency has been used for many years in at-risk pregnancies by means of amniotic fluid hormonal measurement of 17-OH-progesterone.8,143,199,200,201,202,203,204 Genetic diagnosis has now replaced amniotic fluid hormonal measurement. Previously genetic diagnosis was performed in the second trimester by HLA serotyping of fetal cells cultured from the amniotic fluid.205 With few exceptions, a fetus sharing both HLA haplotypes with the affected proband was predicted to be affected. With the advent of chorionic villus sampling (CVS), evaluation of the fetus at risk became possible in the first trimester (9 to 11 weeks'gestation). In early studies, diagnosis was accomplished using cDNA probes for the genes encoding HLA class I and class II antigens, complement C4, and/or 21-hydroxylase.206,207,208,209 Currently the basic approach in molecular diagnosis for direct examination of the 21-hydroxylase genotype of the proband fetus is polymerase chain reaction amplification of the complete sequence for the 21-hydroxylase gene primary transcript from DNA obtained from the CVS biopsy sample.210,211,212,213
Algorithm for Prenatal Diagnosis and Treatment
Before pregnancy in an at-risk family, as much detailed hormonal and genetic information as possible should be obtained on the index case, on any siblings, and on the parents in order to predict the 21-hydroxylase deficiency status of the fetus. The mutations and/or deletions of the 21-hydroxylase genes in the index case and the parents should be identified using the molecular approach just described.
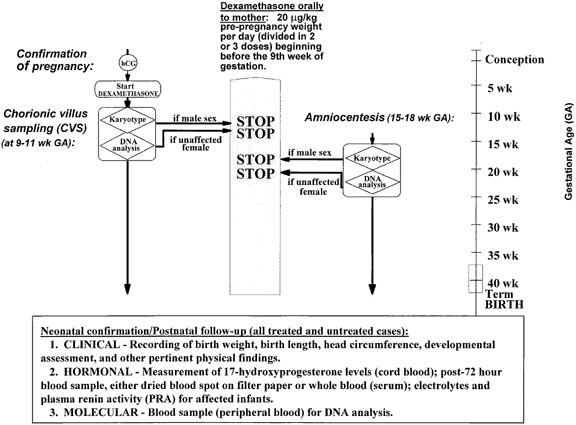
As soon as the pregnancy is confirmed, treatment with dexamethasone (20 mg/kg per day), which at this stage is necessarily blind to the status of the fetus, is started to prevent masculinization in anticipation of an affected genetic female. DNA obtained by CVS at the 9th to 11th weeks is analyzed to search for mutations and/or deletions in the CYP21 gene. Until the results of the biopsy are known, dexamethasone is continued. If the fetus is definitively shown to be unaffected or is a genetic male, dexamethasone may safely be stopped.
If the family is unwilling or unable to participate in CVS, or if the results of the biopsy have been equivocal or contradictory, similar studies at 15 to 18 weeks' gestation on cells obtained by amniocentesis are performed, and the appropriate decisions are made with regard to further management, as outlined earlier. To allow hormonal evaluation of fetal adrenal suppression, amniocentesis may be performed (1) if there is any uncertainty about the status of the fetus based on CVS analysis, and (2) in cases predicted to be affected. Suspending treatment for measurement of amniotic fluid hormones to confirm diagnosis is not advised because prenatal masculinization has been more severe than expected with suspension of treatment for 5 to 7 days before amniocentesis and with cessation of treatment before term. The current recommendation is to continue dexamethasone uninterrupted until delivery (Fig. 6), even if amniocentesis is performed for reasons other than diagnosis.
|
PRENATAL DIAGNOSIS OF 11 β -HYDROXYLASE DEFICIENCY.
Prenatal diagnosis of 11β-hydroxylase deficiency was formerly accomplished by the measurement of 11-deoxycortisol and tetrahydro-11-deoxycortisol in amniotic fluid and of tetrahydro-11-deoxycortisol in maternal urine.214,215 Prenatal diagnosis is now possible for this disease by polymerase chain reaction amplification of exons and selective probing with allele-specific oligonucleotides or by direct sequencing.151
Prenatal Therapy and Current Recommendations
With CVS, which is performed before the critical period of fetal genital differentiation, hormonal treatment can be started in time to reduce significantly or even prevent genital ambiguity in affected female fetuses.
PRENATAL TREATMENT.
Prenatal treatment with dexamethasone has been employed for 15 years in fetuses at risk for 21-hydroxylase deficiency. Theoretically, institution of glucocorticoid therapy at 6 to 7 weeks' gestation and no later than the 9th week, before onset of adrenal androgen secretion, should effectively suppress adrenal androgen production and allow normal separation of the vaginal and urethral orifices, in addition to preventing clitoromegaly.
From 1986 to 1998, prenatal diagnosis and treatment of congenital adrenal hyperplasia caused by 21-OHD was carried out in 403 pregnancies in The New York Presbyterian Hospital-Weill Cornell Medical Center, of which 84 babies were affected with classical 21-OHD.216 Of these, 52 were females, 36 of whom were treated prenatally with dexamethasone. Dexamethasone administered before 10 weeks of gestation was effective in reducing virilization. No significant or enduring side effects were noted in either the mothers (other than greater weight gain in the treated mothers) or the fetuses, indicating that dexamethasone treatment is safe. Prenatally treated newborns did not differ in weight, length, or head circumference from untreated, unaffected newborns.
Currently, the recommended dosage is 20 mg/kg per day of dexamethasone divided in three equal doses, with a maximum daily dose of 1.5 mg. Diagnosis by DNA analysis requires chorionic villus sampling in approximately the 10th week of gestation, or later, in the 2nd trimester, sampling of amniotic fluid cells obtained by amniocentesis. If the fetus is determined to be a male upon karyotype or an unaffected female upon DNA analysis, treatment is discontinued. Otherwise, treatment is continued to term. This is the first instance of an inborn metabolic error to be successfully treated prenatally.
A report has questioned the safety of long-term prenatal glucocorticoid treatment of fetuses potentially affected with congenital adrenal hyperplasia.217 To date, however, no fetus of a mother treated with dexamethasone in low doses has been found to have any congenital malformations other than genital ambiguity in the largest human studies. Specifically, no cases of cleft palate, placental degeneration, or fetal death have been reported, findings that have been observed in a rodent model of in utero exposure to high-dose glucocorticoids.218 Normal birth weight and height, head circumference, as well as normal physical development, have been reported for treated fetuses in the largest human studies.216,219,220
There are no data to suggest that there are any adverse effects of partial prenatal treatment in the case of males or unaffected females. Dexamethasone treatment should be initiated before the 10th week of pregnancy and maintained until diagnosis by CVS and may be further continued until amniocentesis at 16 to 18 weeks, apparently without being deleterious to the fetus. If the risk of either procedure is considered too great by the family, treatment of a possibly unaffected fetus up until term may be an acceptable alternative.
Treatment of affected females prevents potential sex misassignment, repeated genital surgeries that cannot easily recreate natural genital structures, and potential psychological effects. Long-term studies that are currently in progress are needed to conclusively determine outcome of treatment.
ACKNOWLEDGEMENT
Supported by USPHS National Institutes of Health grant HD00072 and grant RR06020 from the General Clinical Research Centers Program, Division of Research Resources of the NIH.
REFERENCES
Jost A: Steroids and sex differentiation o f the mammalian foetus. Excerpta Medica International Congress Series 132: 74, 1996 |
|
Josso N (ed): Differentiation of the Genital Tract: Stimulators and Inhibitors, pp 165–203. New York, Academic Press, 1981 |
|
Wilkins L: Abnormal Sex Differentiation: Hermaphroditism and Gonadal Dysgenesis, pp 297–338. Springfield, IL, Charles C Thomas, 1965 |
|
White PC, New MI, Dupont B: Congenital adrenal hyperplasia (1). N Engl J Med 316: 1519, 1987 |
|
White PC, New MI, Dupont B: Congenital adrenal hyperplasia (2). N Engl J Med 316: 1580, 1987 |
|
New MI, White PC, Pang S et al (eds): The Adrenal Hyperplasias, pp 1881–1917. New York, McGraw-Hill, 1989 |
|
Wilkins L: Adrenal cortex: Virilizing adrenal hyperplasia; virilizing and feminizing tumors; and primary hyperaldosteronism. In Wilkins L (ed): The Diagnosis and Treatment of Endocrine Disorders in Childhood and Adolescence. Springfield, IL, Charles C Thomas, 1965 |
|
New MI, Levine LS: Congenital adrenal hyperplasia. In Harris H, Hirschhorn K (eds): Advances in Human Genetics, Vol 4, pp 251–326. New York, Plenum Publishing, 1973 |
|
Bongiovanni A: Congenital adrenal hyperplasia and related conditions. In Stanbury J, Wyngaarden J, Fredrickson D (eds): The Metabolic Basis of Inherited Disease, p 868. New York, McGraw-Hill, 1978 |
|
Sydnor K, Kelley V, Raile R: Blood adrenocorticotrophin in children with congenital adrenal hyperplasia. Proc Soc Exp Biol Med 82: 695, 1953 |
|
Binoux M, Pham-huu-trung M, Gourmelen M: Plasma ACTH in adrenogenital syndrome. Acta Paediatr Scand 61: 269, 1972 |
|
Ganong W, Alpert L, Lee T: ACTH and the regulation of adrenocortical secretion. N Engl J Med 290: 1006, 1974 |
|
Butler GC, Marrian GF: The isolation of pregnane-3,17,20-triol from the urine of women showing the adrenogenital syndrome. J Biol Chem 119: 565, 1937 |
|
Hughes I, Winter J: The application of a serum 17OH-progesterone radioimmunoassay to the diagnosis and management of congenital adrenal hyperplasia. J Pediatr 88: 766, 1976 |
|
Mason H, Kepler E: Isolation of steroids from urine of patients with adrenal cortical tumors and adrenal cortical hyperplasia: A new 17-ketosteroid, androstane-3a, 11-diol-17-one. J Biol Chem 161: 235, 1945 |
|
Stoner E, Dimartino-Nardi J, Kuhnle U et al: Is salt-wasting in congenital adrenal hyperplasia due to the same gene as the fasciculata defect? Clin Endocrinol (Oxf) 24: 9, 1986 |
|
Luetscher JA: Studies of aldosterone in relation to water and electrolyte balance in man. Recent Prog Horm Res 12: 175, 1956 |
|
Speiser PW, Agdere L, Ueshiba H et al: Aldosterone synthesis in salt-wasting congenital adrenal hyperplasia with complete absence of adrenal 21-hydroxylase. N Engl J Med 324: 145, 1991 |
|
Merke D, Chrousos G, Eisenhofer G et al: Adrenomedullary dysplasia and hypofunction in patients with classic 21-hydroxylase deficiency. N Engl J Med 343: 1362, 2000 |
|
Rosenbloom AL, Smith DW: Varying expression for salt losing in related patients with congenital adrenal hyperplasia. Pediatrics 38: 215, 1966 |
|
Jones H, Jones G: The gynecological aspects of adrenal hyperplasia and allied disorders. Am J Obstet Gynecol 68: 1330, 1954 |
|
Jefferies W, Weir W, Weir D et al: The use of cortisone and related steroids in infertility. Fertil Steril 9: 145, 1958 |
|
Decourt MJ, Jayle MF, Baulieu E: Virilisme cliniquement tardif avec excretion de pregnanetriol et insuffisance de la production du cortisol. Ann Endocrinol (Paris) 18: 416, 1957 |
|
Abraham GE, Corrales PC, Teller RC: Radioimmunoassay of plasma 17-hydroxyprogesterone. Anal Lett 5: 915, 1972 |
|
Rosenwaks Z, Lee PA, Jones GS et al: An attenuated form of congenital virilizing adrenal hyperplasia. J Clin Endocrinol Metab 49: 335, 1979 |
|
Levine LS, Dupont B, Lorenzen F et al: Cryptic 21-hydroxylase deficiency in families of patients with classical congenital adrenal hyperplasia. J Clin Endocrinol Metab 51: 1316, 1980 |
|
Kohn B, Levine LS, Pollack MS et al: Late-onset steroid 21-hydroxylase deficiency: A variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab 55: 817, 1982 |
|
Pollack MS, Levine LS, O'Neill GJ et al: HLA linkage and B14, DR1, BfS haplotype association with the genes for late onset and cryptic 21-hydroxylase deficiency. Am J Hum Genet 33: 540, 1981 |
|
Laron Z, Pollack MS, Zamir R et al: Late-onset 21-hydroxylase deficiency and HLA in the Ashkenazi population: A new allele at the 21-hydroxylase locus. Hum Immunol 1: 55, 1980 |
|
Levine LS, Dupont B, Lorenzen F et al: Genetic and hormonal characterization of cryptic 21-hydroxylase deficiency. J Clin Endocrinol Metab 53: 1193, 1981 |
|
Blankstein J, Faiman C, Reues FI et al: Adult-onset familial adrenal 21-hydroxylase deficiency. Am J Med 68: 441, 1980 |
|
Migeon CJ, Rosenwaks Z, Lee PA et al: The attenuated form of congenital adrenal hyperplasia as an allelic form of 21-hydroxylase deficiency. J Clin Endocrinol Metab 51: 647, 1980 |
|
Speiser PW, Dupont B, Rubinstein P et al: High frequency of nonclassical steroid 21-hydroxylase deficiency. Am J Hum Genet 37: 650, 1985 |
|
New MI, Gertner JM, Speiser PW et al: Growth and final height in classical and nonclassical 21-hydroxylase deficiency. J Endocrinol Invest 12: 91, 1989 |
|
Speiser PW, Serrat J, New MI et al: Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab 75: 1421, 1992 |
|
Pang SY, Lerner AJ, Stoner E et al: Late-onset adrenal steroid 3 beta-hydroxysteroid dehydrogenase deficiency. I. A cause of hirsutism in pubertal and postpubertal women. J Clin Endocrinol Metab 60: 428, 1985 |
|
Gangemi M, Benato M, Guacci A et al: Stimulation tests in adrenogenital syndrome induced by 21-OH deficit. Clin Exp Obstet Gynecol 10: 127, 1983 |
|
Birnbaum M, Rose L: The partial adrenocortical hydroxylase deficiency syndrome in infertile women. Fertil Steril 32: 536, 1979 |
|
Moran C, Azziz R, Carmina E et al: 21-Hydroxylase-deficient nonclassic adrenal hyperplasia is a progressive disorder: A multicenter study. Am J Obstet Gynecol 183: 1468, 2000 |
|
Pang SY, Wallace MA, Hofman L et al: Worldwide experience in newborn screening for classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatrics 81: 866, 1988 |
|
Sherman SL, Aston CE, Morton NE et al: A segregation and linkage study of classical and nonclassical 21-hydroxylase deficiency. Am J Hum Genet 42: 830, 1988 |
|
Zerah M, Ueshiba H, Wood E et al: Prevalence of nonclassical steroid 21-hydroxylase deficiency based on a morning salivary 17-hydroxyprogesterone screening test: A small sample study. J Clin Endocrinol Metab 70: 1662, 1990 |
|
Dupont B, Oberfield SE, Smithwick EM et al: Close genetic linkage between HLA and ongenital adrenal hyperplasia (21-hydroxylase deficiency). Lancet 2: 1309, 1977 |
|
Levine LS, Zachmann M, New MI et al: Genetic mapping of the 21-hydroxylase-deficiency gene within the HLA linkage group. N Engl J Med 299: 911, 1978 |
|
Sinnott PJ, Dyer PA, Price DA et al: 21-hydroxylase deficiency families with HLA identical affected and unaffected sibs. J Med Genet 26: 10, 1989 |
|
Sinnott P, Collier S, Costigan C et al: Genesis by meiotic unequal crossover of a de novo deletion that contributes to steroid 21-hydroxylase deficiency. Proc Natl Acad Sci USA 87: 2107, 1990 |
|
Collier S, Tassabehji M, Sinnott P et al: A de novo pathological point mutation at the 21-hydroxylase locus: Implications for gene conversion in the human genome. Nat Genet 3:260, 1993. Published erratum appears in Nat Genet 4: 101, 1993 |
|
Hejtmancik J, Black S, Harris S et al: Congenital 21-hydroxylase deficiency as a new deletion mutation: Detection in a proband during subsequent prenatal diagnosis by HLA typing and DNA analysis [see comments]. Hum Immunol 35: 246, 1992 |
|
Tajima T, Fujieda K, Fujii-Kuriyama Y: De novo mutation causes steroid 21-hydroxylase deficiency in one family of HLA-identical affected and unaffected siblings. J Clin Endocrinol Metab 77: 86, 1993 |
|
Dupont B, Pollack M, Levine L et al: Congenital adrenal hyperplaisa and HLA: Joint report from the Eighth International Histocompatibility Workshop. In Terasaki P (ed): Histocompatibility Testing, pp 693–706. Los Angeles, UCLA Tissue Typing Laboratory, 1981 |
|
Aston CE, Sherman SL, Morton NE et al: Genetic mapping of the 21-hydroxylase locus: Estimation of small recombination frequencies. Am J Hum Genet 43: 304, 1988 |
|
Klouda P, Harris R, Price D: Linkage and association between HLA and 21-OH deficiency. J Med Genet 17: 337, 1980 |
|
O'Neill GJ, Dupont B, Pollack MS et al: Complement C4 allotypes in congenital adrenal hyperplasia due to 21-hydroxylase deficiency: Further evidence for different allelic variants at the 21-hydroxylase locus. Clin Immunol Immunopathol 23: 312, 1982 |
|
Awdeh Z, Raum D, Yunis E et al: Extended HLA/complement allele haplotypes: Evidence for T/t-like complex in man. Proc Natl Acad Sci USA 80: 259, 1983 |
|
Fleischnick E, Awdeh Z, Raum D et al: Extended MHC haplotypes in 21-hydroxylase-deficiency congenital adrenal hyperplasia: Shared genotypes in unrelated patients. Lancet 1: 152, 1983 |
|
Dupont B, Virdis R, Lerner AJ et al: Distinct HLA-B antigen associations for the salt-wasting and simple virilizing forms of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. In Albert ED, Baur MP, Mayr WR et al (eds): Histocompatibility Testing, p 660. New York, Springer-Verlag, 1984 |
|
Raum D, Awdeh Z, Anderson J et al: Human C4 haplotypes with duplicated C4A or C4B. Am J Hum Genet 36: 72, 1984 |
|
Kominami S, Ochi H, Kobayashi Y et al: Studies on the steroid hydroxylation system in adrenal cortex microsomes: Purification and characterization of cytochrome P-450 specific for steroid C-21 hydroxylation. J Biol Chem 255: 3386, 1980 |
|
Higashi Y, Yoshioka H, Yamane M et al: Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: A pseudogene and a genuine gene. Proc Natl Acad Sci USA 83: 2841, 1986 |
|
White PC, New MI, Dupont B: Structure of the human steroid 21-hydroxylase genes. Proc Natl Acad Sci USA 83: 5111, 1986 |
|
Carroll MC, Campbell RD, Porter RR: The mapping of 21-hydroxylase genes adjacent to complement component C4 genes in HLA, the major histocompatibility complex in man. Proc Natl Acad Sci USA 82: 521, 1985 |
|
White PC, Grossberger D, Onufer BJ et al: Two genes encoding steroid 21-hydroxylase are located near the genes encoding the fourth component of complement in man. Proc Natl Acad Sci USA 82: 1089, 1985 |
|
White PC, Vitek A, Dupont B et al: Characterization of frequent deletions causing steroid 21-hydroxylase deficiency. Proc Natl Acad Sci USA 85: 4436, 1988 |
|
Tusie-Luna M, White P: Gene conversions and unequal crossovers between CYP21 (steroid 21-hydroxylase gene) and CYP21P involve different mechanisms. Proc Natl Acad Sci USA 92: 10796, 1995 |
|
White PC, New MI, Dupont B: HLA-linked congenital adrenal hyperplasia results from a defective gene encoding a cytochrome P-450 specific for steroid 21-hydroxylation. Proc Natl Acad Sci USA 81: 7505, 1984 |
|
Rodrigues NR, Dunham I, Yu CY: Molecular characterization of the HLA-linked steroid 21-hydroxylase B gene from an individual with congenital adrenal hyperplasia. EMBO J 6: 1653, 1987 |
|
Lajic S, Wedell A: An intron 1 splice mutation and a nonsense mutation (W23X) in CYP21 causing severe congenital adrenal hyperplasia. Hum Genet 98: 182, 1996 |
|
Ezquieta B, Oyarzabal M, Jariego C et al: A novel frameshift mutation in the first exon of the 21-OH gene found in homozygosity in an apparently nonconsanguineous family. Horm Res 51: 135, 1999 |
|
Tusie-Luna MT, Speiser PW, Dumic M et al: A mutation (Pro-30 to Leu) in CYP21 represents a potential nonclassic steroid 21-hydroxylase deficiency allele. Mol Endocrinol 5: 685, 1991 |
|
Lajic S, Nikoshkov A, Holst M et al: Effects of missense mutations and deletions on membrane anchoring and enzyme function of human steroid 21-hydroxylase (P450c21). Biochem Biophys Res Commun 257: 384, 1999 |
|
Krone N, Braun A, Roscher A et al: A novel frameshift mutation (141delT) in exon 1 of the 21-hydroxylase gene (CYP21) in a patient with the salt wasting form of congenital adrenal hyperplasia. Hum Mutat 14: 90, 1999 |
|
Lobato M, Ordonez-Sanchez M, Tusie-Luna M et al: Mutation analysis in patients with congenital adrenal hyperplasia in the Spanish population: Identification of putative novel steroid 21-hydroxylase deficiency alleles associated with the classic form of the disease. Hum Hered 49: 169, 1999 |
|
Lee H, Chao H, Lee Y et al: Identification of four novel mutations in the CYP21 gene in congenital adrenal hyperplasia in the Chinese. Hum Genet 103: 304, 1998 |
|
Higashi Y, Tanae A, Inoue H et al: Aberrant splicing and missense mutations cause steroid 21-hydroxylase [P-450(c21)] deficiency in humans: possible gene conversion products. Proc Natl Acad Sci USA 85: 7486, 1988 |
|
Krone N, Roscher A, Schwarz H et al: Comprehensive analytical strategy for mutation screening in 21-hydroxylase deficiency. Clin Chem 44: 2075, 1998 |
|
Wedell A, Ritzen EM, Haglund SB et al: Steroid 21-hydroxylase deficiency: Three additional mutated alleles and establishment of phenotype-genotype relationships of common mutations. Proc Natl Acad Sci USA 89: 7232, 1992 |
|
Witchel S, Smith R, Suda-Hartman M: Identification of CYP21 mutations, one novel, by single strand conformational polymorphism (SSCP) analysis. Hum Mutat 13: 172, 1999 |
|
Amor M, Parker KL, Globerman H et al: Mutation in the CYP21B gene (Ile-172-Asn) causes steroid 21-hydroxylase deficiency. Proc Natl Acad Sci USA 85: 1600, 1988 |
|
Nikoshkov A, Lajic S, Vlamis-Gardikas A et al: Naturally occurring mutants of human steroid 21-hydroxylase (P450c21) pinpoint residues important for enzyme activity and stability. J Biol Chem 273: 6163, 1998 |
|
Speiser PW, New MI, White PC: Molecular genetic analysis of nonclassic steroid 21-hydroxylase deficiency associated with HLA-B14,DR1. N Engl J Med 319: 19, 1988 |
|
Krone N, Braun A, Roscher A et al: Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. J Clin Endocrinol Metab 85: 1059, 2000 |
|
Levo A, Partanen J: Novel nonsense mutation (W302X) in the steroid 21-hydroxylase gene of a Finnish patient with the salt-wasting form of congenital adrenal hyperplasia. Hum Mutat 9: 363, 1997 |
|
Harada F, Kimura A, Iwanaga E et al: Gene conversion-like events cause steroid 21-hydroxylase deficiency in congenital adrenal hyperplasia. Proc Natl Acad Sci USA 84: 8091, 1987 |
|
Wedell A, Luthman H: Steroid 21-hydroxylase deficiency: Two additional mutations in salt-wasting disease and rapid screening of disease-causing mutations. Hum Mol Genet 2: 499, 1993 |
|
Ordonez-Sanchez M, Ramirez-Jimenez S, Lopez-Gutierrez A et al: Molecular genetic analysis of patients carrying steroid 21-hydroxylase deficiency in the Mexican population: Identification of possible new mutations and high prevalence of apparent germ-line mutations. Hum Genet 102: 170, 1998 |
|
Globerman H, Amor M, Parker KL et al: Nonsense mutation causing steroid 21-hydroxylase deficiency. J Clin Invest 82: 139, 1988 |
|
Helmberg A, Tusie-Luna M-T, Tabarelli M. et al: R339H and P453S: CYP21 mutations associated with nonclassic steroid 21-hydroxylase deficiency that are not apparent gene conversions. Mol Endocrinol 6: 1318, 1992 |
|
Chiou SH, Hu MC, Chung BC: A missense mutation at Ile172-Asn or Arg356-Trp causes steroid 21-hydroxylase deficiency. J Biol Chem 265: 3549, 1990 |
|
Lajic S, Levo A, Nikoshkov A et al: A cluster of missense mutations at Arg356 of human steroid 21-hydroxylase may impair redox partner interaction. Hum Genet 99: 704, 1997 |
|
Kirby-Keyser L, Porter C, Donohoue P: E380D: A novel point mutation of CYP21 in an HLA-homozygous patient with salt-losing congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hum Mutat 9: 81, 1997 |
|
Billerbeck A, Bachega T, Frazatto E et al: A novel missense mutation, GLY424SER, in Brazilian patients with 21-hydroxylase deficiency. J Clin Endocrinol Metab 84: 2870, 1999 |
|
Owerbach D, Sherman L, Ballard AL et al: Pro-453 to ser mutation in CYP21 is associated with nonclassic steroid 21-hydroxylase deficiency. Mol Endocrinol 6: 1211, 1992 |
|
Wedell A, Luthman H: Steroid 21-hydroxylase (P450c21): A new allele and spread of mutations through the pseudogene. Hum Genet 91: 236, 1993 |
|
Speiser PW, Dupont J, Zhu D et al: Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest 90: 584, 1992 |
|
Wilson RC, Mercado AB, Cheng KC et al: Steroid 21-hydroxylase deficiency: genotype may not predict phenotype. J Clin Endocrinol Metab 80: 2322, 1995 |
|
Barbat B, Bogyo A, Raux-Demay M et al: Screening of CYP21 gene mutations in 129 French patients affected by steroid 21-hydroxylase deficiency. Hum Mutat 5: 126, 1995 |
|
Morel Y, Bristow J, Gitelman S et al: Transcript encoded on the opposite strand of the human steroid 21-hydroxylase/complement component C4 gene locus. Proc Natl Acad Sci USA 86: 6582, 1989 |
|
Carrera P, Ferrari M, Beccaro F et al: Molecular characterization of 21-hydroxylase deficiency in 70 Italian families. Hum Hered 43: 190, 1993 |
|
Evgrafov O, Polyakov A, Dzenis I et al: Preliminary investigation of mutations in 21-hydroxylase gene in patients with congenital adrenal hyperplasia in Russia. Hum Mutat 5: 131, 1995 |
|
Nikoshkov A, Lajic S, Holst M et al: Synergistic effect of partially inactivating mutations in steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab 82: 194, 1997 |
|
Jaaskelainen J, Levo A, Voutilainen R et al: Population-wide evaluation of disease manifestation in relation to molecular genotype in steroid 21-hydroxylase (CYP21) deficiency: Good correlation in a well-defined population. J Clin Endocrinol Metab 82: 3293, 1997 |
|
Miller W: Phenotypic heterogeneity associated with the splicing mutation in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 82: 1304, 1997 |
|
Bachega T, Billerbeck A, Madureira G et al: Low frequency of CYP2B deletions in Brazilian patients with congenital adrenal hyperplasia due to 21-hydroxylas deficiency. Hum Hered 49: 9, 1999 |
|
Witchel S, Smith R, Crivellaro C et al: CYP21 mutations in Brazilian patients with 21-hydroxylase deficiency. Hum Genet 106: 414, 2000 |
|
Balsamo A, Cacciari E, Baldazzi L et al: CYP21 analysis and phenotype/genotype relationship in the screened population of the Italian Emilia-Romagna region. Clin Endocrinol 53: 117, 2000 |
|
Speiser PW, Dupont J, Zhu D et al: Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest 90: 584, 1992 |
|
Nimkarn S, Cerame B, Wei J et al: Congenital adrenal hyperplasia (21-hydroxylase deficiency) without demonstrable genetic mutations. J Clin Endocrinol Metab 84: 378, 1999 |
|
Pang S, Levine LS, Stoner E et al: Nonsalt-losing congenital adrenal hyperplasia due to 3 beta-hydroxysteroid dehydrogenase deficiency with normal glomerulosa function. J Clin Endocrinol Metab 56: 808, 1983 |
|
Bongiovanni A: The adrenogenital syndrome with deficiency of 3B-hydroxysteroid dehydrogenase. J Clin Invest 41: 2086, 1962 |
|
Kenny F, Reynolds J, Green O: Partial 3B-hydroxysteroid dehydrogenase deficiency in a family with congenital adrenal hyperplasia: evidence for increasing 3B-HSD activity with age. Pediatrics 48: 756, 1971 |
|
Parks GA, Bermudez JA, Anast CS et al: Pubertal boy with the 3-beta-hydroxysteroid dehydrogenase defect. J Clin Endocrinol Metab 33: 269, 1971 |
|
Deperetti E, Forest M, Feit J et al: Endocrine studies in two children with male pseudohermaphroditism due to 3b-hydroxysteroid dehydrogenase defect. In Genazzani A, Thijssen J, Suteri PS (eds): Adrenal Androgens, p 141. New York, Raven Press, 1980 |
|
Zachmann M, Vollmin J, Murset G: An unusual type of congenital adrenal hyperplasia probably due to deficiency of 3B-hydroxysteroid dehydrogenase:Case report of a surviving girl and steroid studies. J Clin Endocrinol Metab 30: 719, 1970 |
|
Cathro M, Birchall K, Mitchell F et al:3b:21dihydroxypregn-5-ene-20-one in urine of normal newborn infants and in third day urine of child with deficiency of 3b-hydroxysteroid dehydrogenase. Arch Dis Child 40: 251, 1965 |
|
Janne O, Perheentupa J, Vihko R: Plasma and urinary steroids in an eight year old boy with 3B-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab 31: 162, 1970 |
|
Schneider G, Genel M, Bongiovanni A: Persistant testicular delta 5,4 isomerase-3B-hydroxysteroid dehyrdrogenase deficiency in the 3B-HSD form of congenital adrenal hyperplasia. J Clin Invest 55: 681, 1975 |
|
Peretti E, Forest M, Feut J et al: Endocrine studies in two children with male pseudohermaphroditism due to 3B-hydroxysteroid dehydrogenase defect. In Genazzani A, Thijssen J, Siiteri P (eds): Adrenal Androgens, p 141. New York, Raven Press, 1980 |
|
Rosenfield R, Rich B, Wolfsdorf J et al: Pubertal presentation of congenital 3 beta-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab 51: 345, 1980 |
|
Heinrich U, Bettendorf M, Vecsei P: Male pseudohermaphroditism caused by nonsalt-losing congenital adrenal hyperplasia due to 3 beta-hydroxysteroid dehydrogenase (3 beta-HSD) deficiency. J Steroid Biochem Mol Biol 45: 83, 1993 |
|
Mendonca B, Russell A, Vasconcelos-Leite M et al: Mutation in 3 beta-hydroxysteroid dehydrogenase type II associated with pseudohermaphroditism in males and premature pubarche or cryptic expression in females. J Mol Endocrinol 12: 119, 1994 |
|
Paula F, Dick-de-Paula I, Pontes A et al: Hyperandrogenism due to 3 beta-hydroxysteroid dehydrogenase deficiency with accessory adrenocortical tissue: A hormonal and metabolic evaluation. Braz J Med Biol Res 27: 1149, 1994 |
|
Russell A, Wallace A, Forest M et al: Mutation in the human gene for 3 beta-hydroxysteroid dehydrogenase type II leading to male pseudohermaphroditism without salt loss. J Mol Endocrinol 12: 225, 1994 |
|
Moisan A, Ricketts M, Tardy V et al: New insight into the molecular basis of 3 beta-hydroxysteroid dehydrogenase deficiency: Identification of eight mutations in the HSD3B2 gene eleven patients from seven new families and comparison of the functional properties of twenty-five mutant enzymes. J Clin Endocrinol Metab 84: 4410, 1999 |
|
Bongiovanni A: Late-onset adrenal hyperplasia [letter]. N Engl J Med 314: 450, 1986 |
|
Bongiovanni AM: Congenital adrenal hyperplasia due to 3β-hydroxysteroid dehydrogenase.In New MI, Levine LS (eds): Adrenal Diseases in Childhood, pp 72–82. Basel, Karger, 1984 |
|
Temeck JW, Pang SY, Nelson C et al: Genetic defects of steroidogenesis in premature pubarche. J Clin Endocrinol Metab 64: 609, 1987 |
|
Mathieson J, Couzinet B, Wekstein-Noel S et al: The incidence of late-onset congenital adrenal hyperplasia due to 3 beta-hydroxysteroid dehydrogenase deficiency among hirsute women. Clin Endocrinol (Oxf) 36: 383, 1992 |
|
Simard J, Rheaume E, Sanchez R et al: Molecular basis of congenital adrenal hyperplasia due to 3 beta-hydroxysteroid dehydrogenase deficiency. Mol Endocrinol 7: 716, 1993 |
|
Chang Y, Kappy M, Iwamoto K et al: Mutations in the type II 3 beta-hydroxysteroid dehydrogenase gene in a patient with classic salt-wasting 3 beta-hydroxysteroid dehydrogenase deficiency congenital adrenal hyperplasia. Pediatr Res 34: 698, 1993 |
|
Simard J, Rheaume E, Leblanc J et al: Congenital adrenal hyperplasia caused by a novel homozygous frameshift mutation 273 delta AA in type II 3 beta-hydroxysteroid dehydrogenase gene (HSD3B2) in three male patients of Afghan/Pakistani origin. Hum Mol Genet 3: 327, 1994 |
|
Rheaume E, Sanchez R, Simard J et al: Molecular basis of congenital adrenal hyperplasia in two siblings with classical nonsalt-losing 3 beta-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab 79: 1012, 1994 |
|
Sanchez R, Mebarki F, Rheaume E et al: Functional characterization of the novel L108W and P186L mutations detected in the type II 3 beta-hydroxysteroid dehydrogenase gene of a male pseudohermaphrodite with congenital adrenal hyperplasia. Hum Mol Genet 3: 1639, 1994 |
|
Alos N, Moisan A, Ward L et al: A novel A10E homozygous mutation in the HSD3B2 gene causing severe salt-wasting 3 beta-hydroxysteroid dehydrogenase deficiency in 46,XX and 46,XY French-Canadians: Evaluation of gonadal function after puberty. J Clin Endocrinol Metab 85: 1968, 2000 |
|
Zerah M, Rheaume E, Mani P et al: No evidence of mutations in the genes for type I and type II 3 beta-hydroxysteroid dehydrogenase (3 beta HSD) in nonclassical 3 beta HSD deficiency. J Clin Endocrinol Metab 79: 1811, 1994 |
|
Chang Y, Wang J, Yang X et al: Molecular basis of the type II 3BHSD gene in a patient with mild non-classic salt-wasting 3B-HSD deficiency congenital adrenal hyperplasia. 75th Annual Meeting of the Endocrine Society, Las Vegas, 1993 |
|
Eberlein WR, Bongiovanni AM: Congenital adrenal hyperplasia with hypertension: Unusual steroid pattern in blood and urine. J Clin Endocrinol Metab 15: 1531, 1955 |
|
Eberlein W, Bongiovanni A: Plasma and urinary corticosteroids in the hypertensive form of congenital adrenal hyperplasia. J Biol Chem 223: 85, 1956 |
|
Werder EA, Siebenmann RE, Knorr-Murset W et al: The incidence of congential adrenal hyperplasia in Switzerland: A survey of patients in 1960 to 1974. Helv Paediatr Acta 35: 5, 1980 |
|
New MI, Nemery RL, Chow DM et al: Low-renin hypertension of childhood. In Biglieri EG, Mantero F, Takeda R et al (eds): The Adrenal and Hypertension: From Cloning to Clinic, pp 323–343. New York, Raven Press, 1989 |
|
Rosler A, Leiberman E, Sack J: Clinical variability of congenital adrenal hyperplaisa due to 11B-hydroxylase deficiency. Horm Res 16: 133, 1982 |
|
Glenthoj A, Nielsen M, Starup J et al: HLA and congenital adrenal hyperplasia due to 11b-hydroxylase deficiency. Tissue Antigens 14: 181, 1979 |
|
Brautbar C, Rosler A, Landau H et al: No linkage between HLA and congenital adrenal hyperplasia due to 11-beta-hydroxylase deficiency[letter]. N Engl J Med 300: 205, 1979 |
|
Pang S, Levine LS, Cederqvist LL et al: Amniotic fluid concentrations of delta 5 and delta 4 steroids in fetuses with congenital adrenal hyperplasia due to 21 hydroxylase deficiency and in anencephalic fetuses. J Clin Endocrinol Metab 51: 223, 1980 |
|
Chua S, Szabo P, Vitek A et al: Cloning of cDNA encoding steroid 11 beta-hydroxylase (P450c11). Proc Natl Acad Sci USA 84: 7193, 1987 |
|
Mornet E, Dupont J, Vitek A et al: Characterization of two genes encoding human steroid 11 beta-hydroxylase (P-450(11) beta). J Biol Chem 264: 20961, 1989 |
|
Wada A, Okamoto M, Nonaka Y et al: Aldosterone biosynthesis by a reconstituted cytochrome P45011 beta system. Biochem Biophys Res Commun 119: 365, 1984 |
|
Curnow K, Tusie-Luna M, Pascoe L et al: The product of the CYP11B2 gene is required for aldosterone biosynthesis in the human adrenal cortex. Mol Endocrinol 5: 1513, 1991 |
|
Ogishima T, Shibata H, Shimada H: Aldosterone synthase cytochrome P450 expressed in the adrenals of patients with primary aldosteronism. J Biol Chem 266: 10731, 1991 |
|
White PC, Dupont J, New MI et al: A mutation in CYP11B1 (Arg-448-His) associated with steroid 11 beta-hydroxylase deficiency in Jews of Moroccan origin. J Clin Invest 87: 1664, 1991 |
|
Helmberg A, Ausserer B, Kofler R: Frame shift by insertion of 2 basepairs in codon 394 of CYP11B1 causes congenital adrenal hyperplasia due to steroid 11 beta-hydroxylase deficiency. J Clin Endocrinol Metab 75: 1278, 1992 |
|
Curnow KM, Slutsker L, Vitek J et al: Mutations in the CYP11B1 gene causing congenital adrenal hyperplasia and hypertension cluster in exons 6, 7, and 8. Proc Natl Acad Sci USA 90: 4552, 1993 |
|
Naiki Y, Kawamoto T, Mitsuuchi Y et al: A nonsense mutation (TGG[Trp116]—>TAG [Stop]) in CYP11B1 causes steroid 11 beta-hydroxylase deficiency. J Clin Endocrinol Metab 77: 1677, 1993 |
|
Skinner C, Rumsby G: Steroid 11 beta-hydroxylase deficiency caused by a five base pair duplication in the CYP11B1 gene. Hum Mol Genet 3: 377, 1994 |
|
Skinner CA, Rumsby G, Honour JW: Single strand conformation polymorphism (SSCP) analysis for the detection of mutations in the CYP11B1 gene. J Clin Endocrinol Metab 81: 2389, 1996 |
|
Geley S, Kapelari K, Johrer K et al: CYP11B1 mutations causing congenital adrenal hyperplasia due to 11 beta-hydroxylase deficiency. J Clin Endocrinol Metab 81: 2896, 1996 |
|
Loidi L, Quinteiro C, Barros F et al: The C494F variant in the CYP11B1 gene is a sequence polymorphism in the Spanish population. J Clin Endocrinol Metab 84: 4749, 1999 |
|
Gabrilove J, Sharma D, Dorfman R: Adrenocortical 11B-hydroxylase deficiency and virilism first manifest in the adult woman. N Engl J Med 272: 1189, 1965 |
|
Newmark D, Dluhy R, Williams G: Partial 11- and 21-hydroxylase deficiencies in hirsute women. Am J Obstet Gynecol 27: 594, 1977 |
|
Cathelineau G, Brerault J, Fiet W: Adrenocortical 11B-hydroxylation defect in adult women with postmenarchial onset of symptoms. J Clin Endocrinol Metab 51: 287, 1980 |
|
Rosler A, Leiberman E: Enzymatic defects of steroidogenesis: 11 beta-hydroxylase deficiency congenital adrenal hyperplasia. In New MI, Levine LS (eds): Adrenal Diseases in Childhood: Pathophysiologic and Clinical Aspects, Vol. 13, pp 47–71. Basel, Karger, 1984 |
|
Hurwitz A, Brautbar C, Milwidsky A: Combined 21- and 11B-hydroxylase deficiency in familial congenital adrenal hyperplasia. J Clin Endocrinol Metab 60: 631, 1985 |
|
Birnbaum M, Rose L: Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol 63: 445, 1984 |
|
Pang S, Levine LS, Lorenzen F et al: Hormonal studies in obligate heterozygotes and siblings of patients with 11 beta-hydroxylase deficiency congenital adrenal hyperplasia. J Clin Endocrinol Metab 50: 586, 1980 |
|
Grumbach M, Ducharme J: The effects of androgens on fetal sexual development: Androgen-induced female pseudohermaphroditism. Fertil Steril 11: 157, 1960 |
|
Haymond M, Weldon V: Female pseudohermaphroditism secondary to a maternal virilizing tumor. J Pediatr 82: 682, 1973 |
|
Murset G, Zachmann M, Prader A: Male external genitalia of a girl caused by a virilizing adrenal tumor in the mother. Acta Endocrinol 65: 627, 1970 |
|
Elterman J, Hagen G: Aldosteronism in pregnancy: Association with virilization of female offspring. South Med J 76: 514, 1983 |
|
Fuller P, Pettigrew I, Pike J et al: An adrenal adenoma causing virilization of mother and infant. Clin Endocrinol 18: 143, 1983 |
|
Kamp JV, Seters AV, Moolenaar A et al: Female pseudohermaphroditism due to an adrenal tumor in the mother. Eur J Pediatr 142: 140, 1984 |
|
O'Leary T, Ooi T, Miller J: Virilization of two siblings by maternal androgen-secreting adrenal adenoma. J Pediatr 109: 840, 1986 |
|
Coslovsky R, Ashkenazy M, Lancet M: Female pseudohermaphroditism with adrenal cortical tumor in adulthood. J Endocrinol Invest 8: 63, 1985 |
|
Kenny F, Hashida Y, Askari A: Virilizing tumors of the adrenal cortex. Am J Dis Child 115: 445, 1968 |
|
Schardein J: Congenital abnormalities and hormones during pregnancy: A clinical review. Teratology 22: 251, 1980 |
|
Bongiovanni A, George A, Grumbach M: Masculinization of the female infant associated with estrogenic therpay alone during gestation. J Clin Endocrinol Metab 19: 1004, 1959 |
|
Melnick S, Cole P, Anderson D et al: Rates and risks of diethylstilbestrol related clear-cell adenocarcinoma of the vagina and cervix. N Engl J Med 316: 514, 1987 |
|
Wilkins L, Lewis R, Klein R et al: The suppression of androgen secretion by cortisone in a case of congenital adrenal hyperplasia. Johns Hopkins Hospital Bulletin 86: 249, 1950 |
|
Bartter F: Adrenogenital syndromes from physiology to chemistry. In Lee P, Lotnick LP, Kowaraski A et al (eds): Congenital Adrenal Hyperplasia, p 9. Baltimore, University Park Press, 1977 |
|
Godard C, Riondel A, Veyrat R: Plasma renin activity and aldosterone secretion in congenital adrenal hyperplasia. Pediatrics 41: 883, 1968 |
|
Simopoulos A, Marshall J, Delea C et al: Studies on the deficiency of the deficiency of 21-hydroxylation in patients with congenital adrenal hyperplasia. J Clin Endocrinol 32: 438, 1971 |
|
Strickland A, Kotchen T: A study of the renin-aldosterone system in congenital adrenal hyperplasia. J Pediatr 81: 962, 1972 |
|
Dillon M: Plasma renin activity and aldosterone concentrations in children: Results in salt-wasting states. Arch Dis Child 50: 330, 1975 |
|
Edwin C, Lanes R, Migeon C: Persistence of the enzymatic block in adolescent patients with salt-losing congenital adrenal hyperplasia. J Pediatr 95: 534, 1979 |
|
Rosler A, Levine LS, Schneider B et al: The interrelationship of sodium balance, plasma renin activity and ACTH in congenital adrenal hyperplasia. J Clin Endocrinol Metab 45: 500, 1977 |
|
Kuhnle U, Rosler A, Pareira JA et al: The effects of long-term normalization of sodium balance on linear growth in disorders with aldosterone deficiency. Acta Endocrinol (Copenh) 102: 577, 1983 |
|
Winter J: Current approaches to the treatment of congenital adrenal hyperplasia [editorial]. J Pediatr 97: 81, 1980 |
|
Korth-Schutz S, Virdis R, Saenger P et al: Serum androgens as a continuing index of adequacy of treatment of congenital adrenal hyperplasia. J Clin Endocrinol Metab 46: 452, 1978 |
|
Golden M, Lippe B, Kaplan S: Management of congenital adrenal hyperplasia using serum dehydroepiandrosterone sulfate and 17-hydroxyprogesterone concentrations. Pediatrics 61: 867, 1978 |
|
Cabrera M, Vogiatzi M, New M: Long term outcome in adult males with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab 86: 3070, 2001 |
|
Zerah M, Pang SY, New MI: Morning salivary 17-hydroxyprogesterone is a useful screening test for nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab 65: 227, 1987 |
|
Quintos J, Vogiatzi M, Harbison M et al: Growth hormone therapy alone or in combination with gonadotropin-releasing hormone analog therapy to improve the height deficit in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab 86: 1511, 2001 |
|
Nihoul-Fekete C: Feminizing genitoplasty in the intersex child. In Josso N (ed): The Intersex Child, Vol. 8, p 247. Basel, Karger, 1981 |
|
Lo J, Schwitzgebel V, Tyrrell J et al: Normal female infants born of mothers with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 84: 930, 1999 |
|
Baker S: Psychological management of intersex children. In Josso N (ed): The Intersex Child, Vol 8, p 261. Basel, Karger, 1981 |
|
Dohler K: The special case of hormonal imprinting, the neonatal influence of sex. Experientia 42: 759, 1986 |
|
Dohler K, Hancke J, Srivastava S: Participation of estrogens in female sexual differentiation of the brain: Neuroanatomical, neuroendocrine, and behavioral evidence. Prog Brain Res 61: 99, 1984 |
|
Price P, Wass J, Griffin J et al: High dose androgen therapy in male pseudohermaphroditism due to 5 alpha-reductase deficiency and disorders of the androgen receptor. J Clin Invest 74: 1496, 1984 |
|
Herdt G, Davidson J: The Sambia “turnim-man” :Sociocultural and clinical aspects of gender formation in male pseudohermaphrodites with 5-alpha-reductase deficiency in Papua New Guinea. Arch Sex Behav 17: 33, 1988 |
|
Money J: Determinants of human gender identity/role. In Money J, Musaph H (eds): Handbook of Sexology, pp 57–79. Amsterdam, Elsevier Science, 1977 |
|
Jeffcoate T, Fleigner J, Russell S et al: Diagnosis of the adrenogenital syndrome before birth. Lancet 2: 553, 1965 |
|
Levine L: Prenatal detection of congenital adrenal hyperplasia. In Milunsky A (ed): Genetic Disorders and the Fetus, pp 369–385. New York, Plenum Press, 1986 |
|
Frasier S, Thorneycroft I, Weiss B et al: Elevated amniotic fluid concentration of 17 alpha-hydroxyprogesterone in congenital adrenal hyperplasia [letter]. J Pediatr 86: 310, 1975 |
|
Nagamani M, McDonough P, Ellegood J et al: Maternal and amniotic fluid 17 alpha-hydroxyprogesterone levels during pregnancy: Diagnosis of congenital adrenal hyperplasia in utero. Am J Obstet Gynecol 130: 791, 1978 |
|
Hughes I, Laurence K: Antenatal diagnosis of congenital adrenal hyperplasia. Lancet 2: 7, 1979 |
|
Hughes I, Laurence K: Prenatal diagnosis of congenital adrenal hyperplasia due to 21-hydroxylase deficiency by amniotic fluid steroid analysis. Prenat Diagn 2: 97, 1982 |
|
Pollack MS, Maurer D, Levine LS et al: Prenatal diagnosis of congenital adrenal hyperplasia (21-hydroxylase deficiency) by HLA typing. Lancet 1: 1107, 1979 |
|
Mornet E, Boue J, Raux-Demay M et al: First trimester prenatal diagnosis of 21-hydroxylase deficiency by linkage analysis to HLA-DNA probes and by 17-hydroxyprogesterone determination. Hum Genet 73: 358, 1986 |
|
Speiser PW, Laforgia N, Kato K et al: First trimester prenatal treatment and molecular genetic diagnosis of congenital adrenal hyperplasia (21-hydroxylase deficiency). J Clin Endocrinol Metab 70: 838, 1990 |
|
Karaviti LP, Mercado AB, Mercado MB et al: Prenatal diagnosis/treatment in families at risk for infants with steroid 21-hydroxylase deficiency (congenital adrenal hyperplasia). J Steroid Biochem Mol Biol 41: 445, 1992 |
|
Forest MG, David M, Morel Y: Prenatal diagnosis and treatment of 21-hydroxylase deficiency. J Steroid Biochem Mol Biol 45: 75, 1993 |
|
Owerbach D, Draznin M, Carpenter R et al: Prenatal diagnosis of 21-hydroxylase deficiency congenital adrenal hyperplasia using the polymerase chain reaction. Hum Genet 89: 109, 1992 |
|
Rumsby G, Honour J, Rodeck C: Prenatal diagnosis of congenital adrenal hyperplasia by direct detection of mutations in the steroid 21-hydroxylase gene. Clin Endocrinol 38: 421, 1993 |
|
Speiser PW, White PC, Dupont J et al: Prenatal diagnosis of congenital adrenal hyperplasia due to 21-hydroxylase deficiency by allele-specific hybridization and Southern blot. Hum Genet 93: 424, 1994 |
|
Wilson RC, Wei JQ, Cheng, KC et al: Rapid DNA analysis by allele-specific PCR for detection of mutations in the steroid 21-hydroxylase gene. J Clin Endocrinol Metab 80: 1635, 1995 |
|
Rosler A, Leiberman E, Rosenmann A: Prenatal diagnosis of 11 beta hydroxylase deficiency congenital adrenal hyperplasia. J Clin Endocrinol Metab 49: 546, 1979 |
|
Schumert Z, Rosenmann A, Landau H et al: 11-Deoxycortisol in amniotic fluid: Prenatal diagnosis of congenital adrenal hyperplasia due to 11 beta-hydroxylase deficiency. Clin Endocrinol 12: 257, 1980 |
|
Carlson AD, Obeid JS, Kanellopoulou N et al: Congenital adrenal hyperplasia: update on prenatal diagnosis and treatment. In Labrie F (ed):Xth International Congress on Hormonal Steroids, Vol 69, pp 19–29. Quebec, Canada, 1999 |
|
Seckl J, Miller W: How safe is long-term prenatal glucocorticoid treatment? JAMA 277: 1077, 1997 |
|
Goldman A: Biochemical mechanism of glucocorticoid and phenytoin-induced cleft palate. Curr Top Dev Biol 19: 217, 1984 |
|
Forest MG, Betuel H, David M: Prenatal treatment in congenital adrenal hyperplasia due to 21-hydroxylase deficiency: Update 88 of the French multicentric study. Endocr Res 15: 277, 1989 |
|
Lajic S, Wedell A, Bui T et al: Long-term somatic follow-up of prenatally treated children with congenital adrenal hyperplasia. J Clin Endocrinol Metab 83: 3872, 1998 |